|
La
supuesta
evolución del
flagelo Sean
D. Pitman, M.D.
Marzo de 2005 - Febrero 2009 |
|
ÍndiceLa supuesta evolución del
flagelo
|
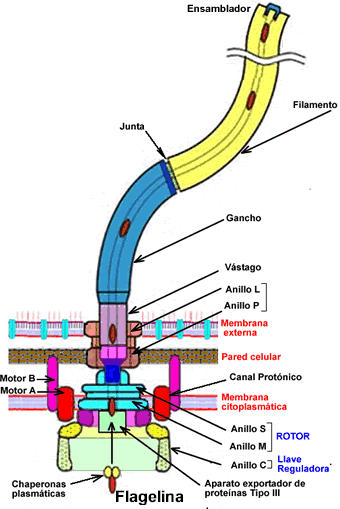
Estructura
del
flagelo bacteriano. La ilustración a la
izquierda (Francis et
al., 1994), es una reconstrucción
promediada en rotación
de micrografías de barrido
electrónico de cuerpos basales
del gancho. Las designaciones de las diversas
partes aparecen en la
ilustración de la derecha, procedente y
adaptada del Grupo de
Nanomáquinas Protónicas (Protonic
NanoMachine
Group) de la Universidad de Osaka.
El designio — ¿mera apariencia?
A
MAYORÍA DE los científicos
modernos creen
que todos los
seres vivos, con todas sus diversas
partes y sistemas
funcionales, evolucionaron por medio de un proceso
de mutaciones al
azar y de
selección natural a partir de una
línea de descendencia
común a lo largo de cientos
de millones de años. Naturalmente, las
famosas observaciones y
documentaciones
que hizo Darwin de diversos cambios reales a
través del tiempo
en muchos
organismos ayudaron a popularizar este concepto.
Desde entonces, las
interpretaciones de la columna geológica y
del registro
fósil, junto con muchos
ejemplos modernos de evolución en tiempo
real, como la
rápida aparición de la resistencia
a los antibióticos en las bacterias, parecen
confirmar la
teoría de la
evolución como algo que es «más
que una
teoría». Sin embargo, todavía
hay aquellos que siguen cuestionando
el potencial creativo de un proceso tan carente de
propósito.
¿Puede este
mecanismo aparentemente simple de mutaciones al azar
y la
selección natural
originar realmente la asombrosa complejidad y
diversidad de todos los
seres
vivientes? Para muchos, las maravillas del mundo
natural y del
universo,
especialmente cuando se trata de los seres vivos,
les inspiran tal
maravilla
que intuyen que tienen que haber sido dispuestas
deliberadamente por
una
inteligencia extraordinariamente brillante —por
Dios, o al menos por
una
inteligencia divina. Consideremos una
observación que hace
Keiichi Namba
(director del programa de un equipo de
científicos dedicados a
investigar los
detalles de las diversas etapas del montaje del
flagelo): Una
enorme cantidad de estas macromoléculas
desempeñan cada
papel precisamente como máquinas
diseñadas a
propósito y mantienen las complejas
actividades del sistema.12 El
mecanismo evolutivo
El famoso biólogo materialista británico, Richard Dawkins, describe este proceso como «Escalando el Monte Improbable» en un popular libro que lleva este título. En este libro, Dawkins explica que aunque pueda parecer muy improbable que la montaña de complejidad que existe en la actualidad haya surgido gracias a un azar ciego, la evolución no es un proceso de cambio puramente al azar. La evolución emplea azar para crear pequeños pasos, cada uno de los cuales es bien bueno o malo, o neutral, con respecto a la capacidad reproductiva global del organismo. La Naturaleza, por medio de un proceso que se conoce popularmente como la «supervivencia de los más aptos», da una capacidad de reproducción mayor a aquellos organismos que adquieren cambios positivos, y resta capacidad reproductiva a aquellas criaturas que soportan cambios perjudiciales. Es evidente, entonces, que la siguiente generación quedará más poblada por aquellos organismos con los cambios más beneficiosos. De esta manera, los cambios buenos se acumulan con el paso del tiempo en cada generación, y esta montaña de enorme complejidad va siendo escalada paso a paso, uno por uno.
Discontinuidades
no beneficiosas
Todo esto parece estar muy
bien, y realmente bastante
convincente, excepto quizá por un
pequeño problema. La
selección natural está
limitada en cuanto a que solo puede seleccionar de
una forma positiva
aquellos
cambios funcionales que manifiestan una mejora en la
función
sobre lo que había antes. Pero resulta que
muchos cambios
mutacionales (esto es, cambios en los códigos
genéticos
subyacentes del ADN que
dictan como se forma un organismo en cada uno de sus
detalles) no
tienen en
absoluto ningún efecto en la función
del organismo. Estos
cambios, o
mutaciones, son designados como
«neutros» con respecto a la
selección de Una diferencia neutra puede ser cuestión de «deletrear» de manera diferente el código para la misma función. Esta diferente grafía sigue resultando en la producción de un resultado idéntico, igual o equivalente (como acabo de hacer yo al usar tres palabras diferentes que son prácticamente sinónimas). O bien, puede que existan diferencias neutras entre secuencias igualmente carentes de significado — como la diferencia entre quiziligook y quiziliguck. Ambas carecen igualmente de significado cuando se pronuncian en la mayor parte de situaciones. Por ello, ninguna de las dos podría adquirir una mejor «aptitud» o un mayor significado en un medio determinado que en otro. Es evidente, así, que la selección entre ambas sería idéntica o «neutra» con respecto a la función —es decir, completamente al azar. ¿Por qué debe esto ser un problema para la evolución? La cuestión es que a niveles muy bajos de complejidad funcional (esto es, funciones que precisen de una secuencia muy corta de paisaje genético bien especificada que deba materializarse) la proporción de las secuencias potencialmente benéficas a las secuencias no benéficas es bastante elevada. De modo que la cantidad de diferencias neutras entre una secuencia benéfica y la siguiente secuencia más cercana potencialmente benéfica es relativamente baja. Por ejemplo, consideremos la siguiente secuencia de vocablos en inglés: cat - hat - bat - bad - big - dig - dog. Es fácil pasar por la secuencia de cada tres caracteres en el sistema de la lengua inglesa, debido a que la relación entre términos con significado y sin significado en el «espacio de secuencia» de secuencias de 3 caracteres es de aproximadamente sólo 1 en 18. Sin embargo, esta relación disminuye espectacularmente, de hecho exponencialmente, con cada incremento en longitud mínima de secuencia. Para un espacio de secuencia de 7 caracteres, la proporción es de alrededor de 1 entre 250.000, y esto sin tener siquiera en cuenta la naturaleza «benéfica» de una secuencia determinada en relación con un medio ambiente o situación particulares. Con todo, las secuencias de 7 caracteres están por lo general muy interconectadas, como una red hecha de estrechos caminos entrelazados que rodean grandes espacios de potenciales secuencias carentes de sentido y no beneficiosas. Sin embargo, la disminución exponencial en la proporción se hace evidente, y sus implicaciones son claras. Para funciones de nivel más y más elevado que necesiten secuencias más y más largas con una buena especificación para codificarlas, la proporción entre secuencias con significado y secuencias sin significado llega a hacerse tan pequeña tan rápidamente que cuando se necesita de más de unas pocas docenas de caracteres los caminos y puentes de interconexión que vinculan diversos grupos de islas de secuencias benéficas comienzan a romperse. A unos niveles sorprendentemente bajos de complejidad funcional este proceso aísla las diminutas islas de secuencias benéficas entre sí hasta tal magnitud que simplemente no hay forma de alcanzar estas diminutas islas separadas excepto atravesando el vacío de secuencias no benéficas mediante un proceso de cambio(s) por puro azar con el transcurso del tiempo. Con cada larguero adicional al subir por la escala de la complejidad funcional, este vacío se va haciendo más y más amplio, de forma exponencial, hasta que es simplemente imposible de cruzar por más que añadamos billones de billones de años de tiempo medio. La selección natural es sencillamente ciega cuando se trata de salvar tales vacíos. Sin la guía de la selección natural, salvar estos espacios demanda cantidades exorbitantemente mucho mayores aún, por cuanto las secuencias basura no benéficas de espacio de secuencia tienen que ser eliminadas al azar antes que se descubra una secuencia benéfica muy rara por un golpe de increíble fortuna (véase cálculos en el apéndice). Naturalmente, algunos han sugerido que una sola mutación de inserción, compuesta de la secuencia precisamente idónea de múltiples caracteres, podría cruzar un vacío de consideración entre una isla funcional y otra isla muy alejada. Claro que es cierto, pero el problema aquí es que no servirá cualquier secuencia o inserción de caracteres múltiples. Esta secuencia tiene que ser justamente la idónea para que funcione para muchos tipos de funciones de alto nivel. La probabilidad de que aparezca una secuencia específica de esta clase es tan sumamente remota que estamos hablando de billones de años cuando el vacío llega a tamaños de sólo unas pocas docenas de diferencias de caracteres no beneficiosos. Entonces, incluso si la secuencia necesaria apareciera porque sí en el genoma, se tendría que insertar en el lugar justo y preciso para que funcionase de forma benéfica para una función particular en evolución. La inmensa mayoría de potenciales posiciones de inserción serían perjudiciales o como mucho neutras con respecto a la función global. Así,
queda claro que
conseguir «el resultado
justo» no es una cuestión sencilla.
Como media, se
precisaría literalmente de
billones de billones de años por pruebas al
azar para cruzar
vacíos no
benéficos relativamente pequeños.
Así es, en
resumen, como se nos presenta el
problema. ¿Y cómo se puede resolver? El
flagelo eucariota
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||



Función |
|
|
Motor |
MotA,
MotB,
FliG (C-term) |
|
Base |
FliF,
FliG
(N-term), FliM/N |
|
Maquinaria de exportación |
FlhB, FliQ, FliR, FliP, FliI, FlhA |
|
Eje de transmisión |
FlgB, FlgC, FlgG, FliE |
|
Gancho y adaptadores |
FlgE, FlgL, FlgK, FlgD |
|
Filamento |
FliC, FliD |
Parece que estos 21 genes están al menos cerca del mínimo imprescindible para una función flagelar útil. En combinación con los otros genes necesarios para ayudar en la construcción de la estructura flagelar, el mínimo imprescindible parece seguir estando alrededor de 35 a 40 genes específicos. Así, queda claro que el sistema de motilidad flagelar es muy complejo desde el criterio de la información. Para conseguir la función de la motilidad el flagelo exige un mínimo de varios miles de residuos de aminoácidos que actúan juntos en una disposición muy específica o «especificada» en mutua relación..
Son muchos los intentos que se han hecho para explicar la evolución paso a paso de un sistema tan evidentemente complejo. La mayoría de estas explicaciones son muy superficiales, saltando, con un gesto displicente, sobre enormes vacíos evolutivos que involucran grandes cambios de múltiples proteínas. Sin embargo, ha habido algunos intentos más meritorios. Quizá uno de los mejores intentos para tratar de explicar la evolución del flagelo es la proposición de Nicholas J. Matzke en este artículo de 2003: «Evolution in (Brownian) space: a model for the origin of the bacterial flagellum [La evolución en el espacio (Browniano): un modelo para el origen del flagelo bacteriano]».
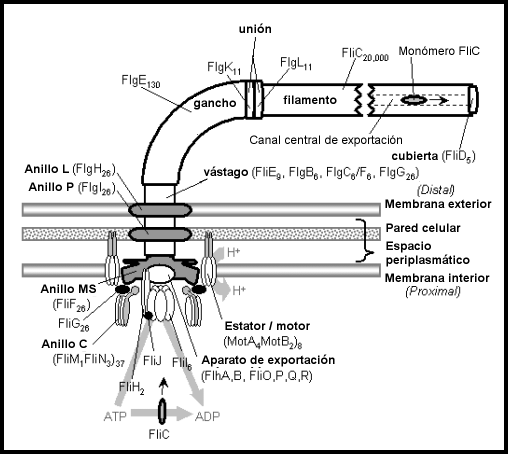
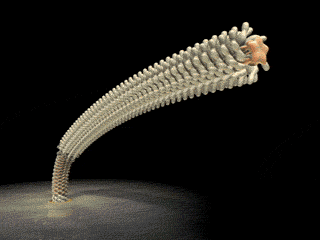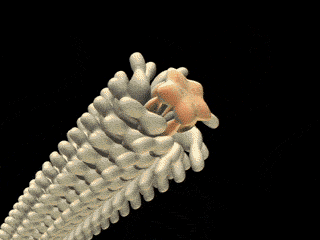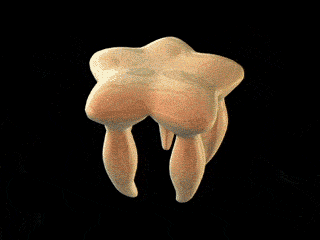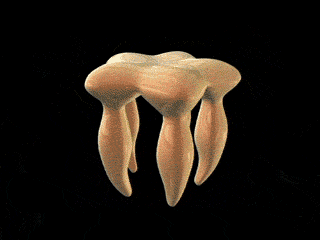
Sección de un flagelo bacteriano
típico.
Las subestructuras se designan
con negritas, y las
proteínas en
tipo normal.
Véanse las
tablas al
final.
con negritas, y las proteínas en tipo normal.
Véanse las tablas al final.
El punto de partida
Es cosa extraña que
los evolucionistas propongan
tan generalmente el sistema SSTT como el punto de
partida más
probable, siendo
que se supone que el sistema SSTT habría
evolucionado cientos de
millones de
años después de la
evolución del
flagelo. Sí, la realidad es que varios
científicos han
sugerido en artículos
bastante recientes que hay buenas razones para creer
que el punto de
partida
del SSTT fue a partir del flagelo
plenamente formado, y no al revés.2-7
Se debe
considerar que el
flagelo bacteriano se encuentra tanto en las bacterias
mesofílicas como termofílicas,
Gram-positivas, Gram-negativas y espiroquetas,
mientras que los
sistemas SSTT
están limitados a algunas bacterias
Gram-negativas. No solo
están los sistemas
SSTT restringidos a las bacterias Gram-negativas, sino
también a
bacterias
Gram-negativas patogénicas que atacan de forma
específica
a animales y a
plantas ... ¡que supuestamente evolucionaron
miles de millones de
años después que
hubiera evolucionado la
motilidad flagelar! Además, cuando los genes de
los SSTT se
encuentran en los
cromosomas de las bacterias, su contenido en GC
(guanina/citosina) es
generalmente inferior al contenido en GC del genoma
alrededor. Dado que
los
genes de los SSTT se encuentran comúnmente en
plásmidos
de gran virulencia (que
pueden pasar fácilmente entre diferentes
bacterias), esta es
buena evidencia de
la transferencia lateral para explicar la
distribución de los
genes de los
SSTT. Los genes flagelares, en cambio, están
generalmente
repartidos entre
alrededor de 14 operones, no se encuentran en plásmidos,
y su contenido en GC es el mismo
que el del genoma a su alrededor, lo que sugiere que el
código para el flagelo no se ha difundido
mediante transferencia
lateral.
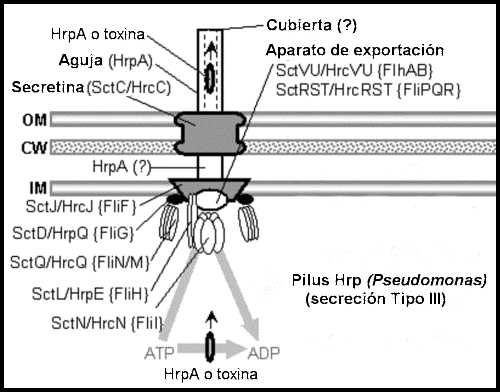
Así que, en todo caso, más bien parece que el sistema SSTT hubiera podido evolucionar procedente del flagelo (que de hecho contiene subcomponentes correspondientes al sistema SSTT, como un cuerpo basal que secreta diversas proteínas no flagelares — incluyendo factores de virulencia), y no a la inversa.
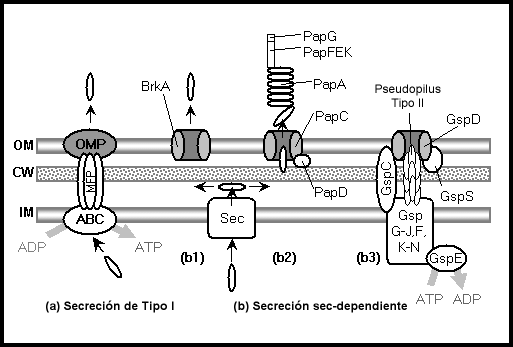
Sistemas de secreción. Las
subestructuras
se designan
con negritas, y las
proteínas en
tipo normal.
Véanse las
tablas al
final.
Una adicional evidencia de esto
procede del hecho de que el
sistema
SSTT exhibe poca
homología con ningún otro sistema
de transporte bacteriano (al menos con los 14
principales). En cambio,
se
supone que la evolución va construyendo a
partir de aquello que
ya existe. Por
cuanto el sistema SSTT es el más complejo de
todos éstos,
¿por qué no
evolucionó a partir de uno de estos sistemas
menos complejos,
manteniendo con
ello algún mayor grado de homología con
al menos uno de
ellos? Estos datos sugieren
que en la «era pre-flagelar» no
existía el sistema
SSTT, ni nada homólogo al
mismo. Por ello, debe haber surgido del flagelo
plenamente formado
mediante la eliminación de
piezas
preexistentes, y
no al revés. Como se ha mencionado, varios
científicos
han estado proponiendo
esta idea en la literatura reciente.2-7 Por
ejemplo,
consideremos el
siguiente artículo publicado en 2008 por Toft y
Fares:
El
encogimiento del genoma es un
rasgo común de la
mayoría de los patógenos y simbiontes
intracelulares. La
reducción de los
tamaños del genoma se encuentra entre las
vías evolutivas
mejor caracterizadas
de los organismos intracelulares para ahorrar y evitar
mantener unos
costosos
procesos biológicos redundantes. Las bacterias
endosimbióticas de los insectos
son ejemplos de economía biológica
llevados a su punto
óptimo debido a que sus
genomas quedan espectacularmente reducidos. Estas
bacterias carecen de
motilidad, y sus procesos bioquímicos
están
íntimamente relacionados con los de
su hospedador. Debido a esta relación, muchos
de los procesos en
estas
bacterias o bien se han perdido o bien han
experimentado un enorme
remodelado
para adaptarse al estilo de vida simbiótico
intracelular. Un
ejemplo de estos
cambios es la estructura flagelar esencial para la
motilidad y
capacidad
infectiva de las bacterias. Nuestro análisis
indica que los
genes responsables
para el montaje del flagelo se han perdido en parte o
en todo en la
mayoría de
los simbiontes intracelulares de las
Gamma-Proteobacterias.
Análisis genómicos comparados indican
que se
han perdido diferencialmente genes flagelares en
bacterias endosimbióticas de insectos. En la
mayoría de los
endosimbiontes sólo se han conservado las
proteínas
implicadas en la
exportación de proteínas dentro de la
ruta del montaje
del flagelo (sistema de
secreción tipo III y el cuerpo basal), mientras
que las
involucradas en la
construcción del filamento y del gancho del
flagelo sólo
se han conservado en
pocos casos, lo que indica un cambio en el
propósito funcional
de esta ruta. En
algunos endosimbiontes, los genes que controlan el
interruptor de la
exportación de proteínas y la longitud
del gancho han
experimentado una
divergencia funcional como se constata mediante un
análisis de
su dinámica
evolutiva. Basados en nuestros resultados, sugerimos
que los genes del
flagelo
han divergido funcionalmente para especializarse en la
exportación de proteínas
desde la bacteria hacia el hospedador.13
Pero claro, es muy práctico comenzar la explicación de un sistema de gran complejidad comenzando por el medio —o esto parecería a primera vista. De las alrededor de 27 piezas proteínicas empleadas en la estructura flagelar, 10 de ellas son homólogas con las proteínas en el SSTT. Una de estas 10 es la proteína «FliI». La FliI es una ATPasa anclada a la superficie citoplasmática de la membrana interior y probablemente suministra energía para la síntesis de la maquinaria de exportación o de transporte de proteínas segregadas, que son capturadas de forma selectiva desde el citoplasma para fines de transporte. Luego hay las proteínas que componen el aparato de transporte de la membrana interior y que probablemente constituyen el canal de conducción de proteínas. Estas incluyen FlhA, FliP, FliQ, FliR, y FlhB. El homólogo flagelar del anillo MS está compuesto de FliF y el homólogo del anillo C está compuesto de FliN y FliG. La última proteína, FliH, tiene una función desconocida.
Parece que la mayor parte de estos 10 homólogos flagelares son necesarios para la función del SSTT. De modo que la suposición de un sistema intacto proto-SSTT es un buen comienzo para tratar de explicar la evolución flagelar. La realidad es que el sistema SSTT es sumamente complejo por derecho propio, y esto sólo se acumula al concepto de que el sistema SSTT no evolucionó a partir de un sistema de menor complejidad, sino que surgió de un sistema de mucha mayor complejidad (el flagelo plenamente formado) por medio de un proceso de eliminación de componentes preexistentes —no por la adición de nuevos componentes. Evidentemente, es mucho más fácil quitar componentes y mantener funciones de un nivel inferior que ya están allí que añadir nuevos componentes a funciones de bajo nivel para conseguir funciones beneficiosas de alto nivel que todavía no existen.
Añádase a
esto
que algunos de los homólogos entre
los sistemas flagelar y SSTT no son tan
homólogos.
En resumen, es muy difícil de explicar la función del sistema SSTT mismo usando mecanismos evolutivos sin dirección ni propósito. Todavía me queda por ver un intento razonable de explicar cómo pudo haberse originado por evolución un sistema SSTT con vacíos neutros lo suficientemente pequeños para ser salvados por mutaciones al azar del tipo que sea.
Matzke y otros
evolucionistas
abordan estos
problemas sugiriendo que algún día
pudiera encontrarse
algún homólogo todavía
no descubierto del aparato secretor del flagelo.
Matzke explica:
Si los sistemas de virulencia de tipo III derivan de flagelos, ¿cuál es la base para emitir la hipótesis de un sistema de secreción de tipo III que sea ancestro de los flagelos? La cuestión quedaría resuelta si se descubrieran homólogos no flagelares del aparato de exportación del tipo III en otras filas bacterianas, que realizasen funciones útiles en el mundo pre-eucariota. Que no se haya descubierto todavía tal cosa constituye un argumento válido en contra del presente modelo, pero al mismo tiempo sirve de predicción: el modelo quedará considerablemente fortalecido si se descubre dicho homólogo. Por el momento, es bastante fácil explicar la falta de descubrimiento de tal homólogo en base a la falta de datos.1
¿De modo que por ahora la evidencia para la evolución del primer paso en la síntesis flagelar está escondida y segura detrás de una «falta de datos»? ¿Dónde está la «detallada» explicación de la evolución flagelar en todo esto? Bien, Matzke y otros imaginan lo que hubiera podido suceder para lograr la evolución del primer sistema proto-SSTT. El origen de este sistema proto-SSTT comienza con un homólogo de FliF, un complejo de porinas de membrana. FlhB, el complejo proteínico que controla el tipo de proteínas secretadas a través del poro, se une de alguna manera a FliF. FlhA, de función desconocida, se añadió también para que junto con FlhB, se pudiera transformar un poro transportador pasivo general en un transportador de sustrato específico. No se sugiere de dónde pudieron proceder FlhB o FlhA ni qué otras funciones hubieran podido tener ni tampoco queda claro cómo sus capacidades selectivas hubieran podido ser de ayuda, especialmente en el caso de que se hubieran seleccionado unas proteínas desacertadas para su transporte.
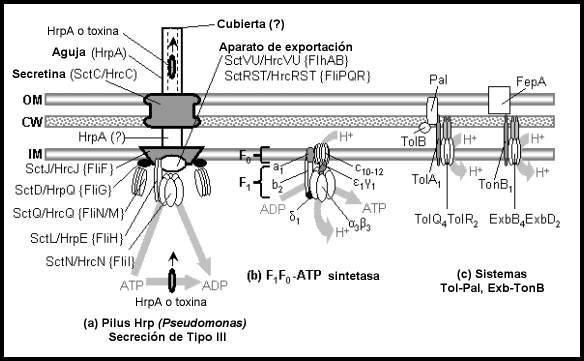
Sistemas con componentes
homólogos a
componentes flagelares.
Las subestructuras se designan
con negritas, y las proteínas en tipo normal.
Véanse las
tablas al
final.
En todo caso, una vez se han combinado la FlhB y FlhA con la FliF, se precisa de energía para el transporte activo. Y aquí entra FliI al rescate. La propuesta es que la F1-αβ ATPasa, un heterohexámero compuesto de subunidades α (no catalíticas) y de subunidades β (catalíticas) que se encuentra en muchos tipos de bacteria, evolucionó a partir de un antecesor común de FliI (un homohexámero compuesto de subunidades catalíticas y fuente de energía para el SSTT), por cuanto FliI comparte ~30% de homología con
Naturalmente, si se
concede que
se pueda obtener
FliI, es fácil conseguir que la fuente de
energía de la
FliI se conecte al poro
del FliF —¿verdad? No
tan
fácil. La FliI
no puede acoplarse directamente a FliF. Se precisa de
otra
proteína llamada
«FliH» para conseguir que
Así que para que
el SSTT
pueda conseguir la
exportación activa de la proteína, se
tienen que disponer
tres complejos
proteínicos de forma precisa (FliI, FliH, y
FliJ) — y esto
simplemente para la
versión imaginaria. Las otras partes del
aparato secretor,
FliOPQR, ni son
consideradas en el «detallado» modelo por
etapas de Matzke
de evolución
flagelar, debido a la «falta de datos».
Además de lo anterior, ¿qué hay del argumento de que las semejanzas entre las proteínas de la F1F0-ATP sintetasa y del aparato exportador flagelar de tipo III respaldan el concepto de que comparten un antecesor primitivo común? Prácticamente inmediatamente, Matzke añade: «Individual-mente, las semejanzas que se citan son fácilmente atribuibles al azar, pero juntas son al menos sugestivas».1 A mi parecer, esto suena a que hay algunos vacíos bastante grandes al menos potencialmente presentes ya en la ruta que se ha pro-puesto. Al menos, estos vacíos no se consideran con ningún detalle en el modelo de Matzke ni en ningún otro modelo que yo conozca. Estas etapas, que parecen exigir cientos de diferencias genéticas bastante específicas, se pasan por alto, simplemente con un gesto displicente, para llegar a la siguiente conclusión:
El
acontecimiento clave en el
origen de la
exportación de tipo III fue la
asociación de una
primitiva F1F0-ATP
sintetasa con una proto-FlhA o FlhB en el interior del
anillo
proto-FliF, convirtiéndolo
de un transportador pasivo a otro activo. Por cuanto
es poco lo que se
conoce
de los detalles de la actividad acopladora de la
ATPasa con la
exportación
proteínica en la exportación de Tipo
III, este paso sigue
siendo especulativo.1
Complejidad
irreducible
Un sistema unitario compuesto de diversas piezas que interaccionan y contribuyen a la función básica, y donde la eliminación de cualquiera de las piezas hace que dicho sistema deje realmente de funcionar.16
Los científicos de la corriente dominante consideran que este argumento ha sido en gran medida refutado a la luz de una investigación bastante reciente. Observan que se ha descubierto que el cuerpo basal del flagelo es similar al sistema de secreción de Tipo III (SSTT), una estructura semejante a una aguja que usan los microorganismos patogénicos como la salmonella para inyectar toxinas en células eucariotas vivas. La base de la aguja tiene muchos elementos en común con el flagelo, pero carece de la mayoría de las proteínas que hacen funcionar a un flagelo. Así, este sistema parece refutar la aseveración de que la eliminación de cualquiera de las piezas del flagelo lo convertiría en inservible. Esto ha llevado a [Kenneth] Miller a observar que «Las piezas de este sistema complejo supuestamente irreducible tienen en realidad funciones propias».17,18
Ahora bien, uno podría construir sistemas funcionales de alto nivel, sistemas que exigen más y más requisitos estructurales mínimos necesarios, con el uso de sistemas preestablecidos más pequeños ya disponibles. Sin embargo, esta potencialidad no elimina la realidad de que los sistemas de más alto nivel tienen un mayor tamaño mínimo y mayores requisitos de especificidad antes que puedan hacerse realidad —incluso en el mínimo sentido. Todos los tipos de funciones tienen sus propios requisitos mínimos. Estos requisitos mínimos no son todos iguales. Y es esta diferencia en los requisitos mínimos lo que los distingue.
La verdadera cuestión es: ¿pueden construirse sistemas irreducibles usando componentes ya presentes en el fondo genético? Y, en tal caso, ¿es igualmente probable acabar con funciones a diferentes niveles de requisitos de tamaño y de especificidad?
Mantengo que los sistemas funcionales que exigen un mínimo de sólo unas pocas docenas de residuos de aminoácidos en una orientación bastante específica pueden evolucionar en un plazo relativamente breve (sólo unas pocas generaciones para una colonia de unos cuantos miles de millones de bacterias). Sin embargo, la probabilidad de que se puedan conseguir funciones de niveles progresivamente más elevados dentro de un plazo breve de tiempo disminuye exponencialmente con cada etapa arriba de esta escala de complejidad funcional irreducible.
Esta noción está respaldada en la literatura. Hay multitud de ejemplos de evolución «en acción» cuando se trata de funciones que exigen un mínimo de sólo unas cuantas docenas de residuos o si las posiciones de los residuos no tienen que ser demasiado especificadas (resistencia a los antibióticos, especificidad mejorada del sistema inmune, capacidad infectiva de fagos, etc.). Sin embargo, cuando se trata de funciones que exigen un mínimo de unos cuantos cientos de residuos bastante especificados operando de manera conjunta y simultánea (como en enzimas de proteínas únicas como la lactasa, la nilonasa, etc.), el número de ejemplos cae espectacularmente y la cantidad de fondos genéticos bacterianos capaces de evolucionar funciones a este nivel, incluso en un medio sumamente selectivo, cae también exponencialmente.
Cuando se llega al nivel de funciones que exigen meramente 1.000 residuos bastante especificados operando conjunta y simultáneamente, simplemente no hay ejemplos de evolución «en acción» mencionados en la literatura —ninguno en absoluto. Todo lo que tenemos al llegar a este punto son historias acerca de cómo los mecanismos evolutivos de mutación al azar y de selección basada en la función tienen que haber hecho la tarea. Es decir, simplemente cuentos basados en nada más que suposiciones. No hay observaciones reales de evolución en acción más allá de este punto —ni un solo ejemplo. Tampoco hay intentos serios de calcular las probabilidades de que la evolución se dé a tales niveles en el período propuesto de unos pocos miles de millones de años durante los que se supone que ha tenido lugar la evolución de la vida sobre esta Tierra.
El trabajo que estamos considerando aquí, de Matzke, no es una excepción. Matzke ni siquiera intenta calcular las probabilidades de que la evolución cruce ninguna de sus propuestas etapas en la ruta de la evolución del flagelo. Sencillamente se apoya, como lo hacen los demás científicos de la línea convencional, en la noción de que las semejanzas de secuencia sólo podrían ser resultado de una relación evolutiva. Los cálculos estadísticos relativos a la capacidad de mutaciones aleatorias y de la selección basada en la función para realmente poder recorrer estas etapas que se proponen parecen simplemente ser innecesarios para los científicos de la línea convencional. ¿Para qué calcular las probabilidades cuando el cuento es tan bonito?
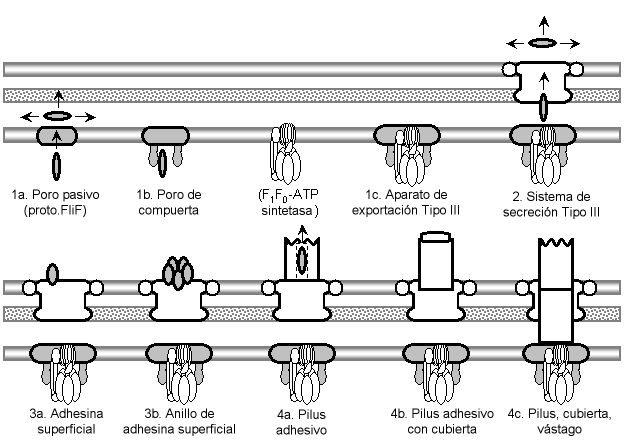
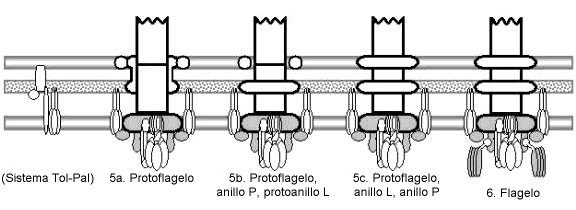
Resumen de Maztke del modelo evolutivo para el origen del flagelo, exhibiendo las seis etapas principales y los intermedios clave. Los componentes blancos tienen homólogos no flagelares identificados o razonablemente probables; los componentes grises tienen homólogos o bien sugeridos pero sin evidencia, o ningún homólogo identificado específico, aunque se pueden postular funciones ancestrales. El modelo comienza con un poro de membrana interna pasivo algo general (1a) que se convierte a un poro para sustrato más específico (1b) por unión de la proto-FlhA y/o FlhB con FliF. La interacción de una F1F0-ATP sintetasa con FlhA/B produce un transportador activo, un primitivo aparato de exportación de tipo III (1c). La adición de una secretina que se asocia con el anillo citoplasmático convierte esto a un sistema de secreción de tipo III (2). Un sustrato mutado de secreción se transforma en una adhesina secretada (o, alterna-tivamente se coopta una adhesina por transposición de la secuencia de reconocimiento de la secreción), y una mutación posterior permite que se una al lado exterior de la secretina (3a). La oligomerización de la adhesina produce un anillo pentamérico, lo que permite más adhesinas superficiales sin que se bloqueen otros sustratos de secreción (3b). La polimerización de este anillo produce un tubo, un pilus primitivo de tipo III (4a; en el diagrama, aparece una estructura axial blanca en lugar de las subunidades individuales de pilina; todas las otras proteínas axiales descienden de esta pilina ancestral común). La oligomerización de una pilina produce la cubierta, que aumenta la velocidad y la eficiencia del montaje (4b). Una pilina duplicada que pierde sus dominios exteriores pasa a ser la proteína del protovástago, que se extiende hacia abajo a través de la secretina y que fortalece la unión del pilus mediante asociación con la base (4c). Adicionales duplicaciones de las proteínas del protovástago, del filamento y de la cubierta, que ocurren antes y después del origen del flagelo (6) producen el resto de las proteínas axiales; estos repetidos eventos de subfuncionalización no se muestran aquí. El protoflagelo (5a) se produce por cooptación de los homólogos de TolQR a partir de un sistema parejo al Tol-Pal; quizá una porción de un homólogo de TolA unido a FliF para producir proto-FliG. A fin de conseguir la mejora de la rotación, la secretina pierde sus sitios de unión al filamento axial, transformándose en el protoanillo P, y el papel de poro de la membrana exterior es asumido por el anillo chaperona de lipoproteína de la secretina, que pasa a ser el protoanillo L (5b). El perfeccionamiento del anillo L y la adición del dominio de muramidasa de la cubierta de FlgJ del vástago (que elimina la necesidad de encontrar una abertura natural en la pared de la célula) resulta en 5c. Finalmente, la unión de una proto-FliN mutante (probablemente un receptor CheC) a FliG acopla el sistema de transducción de señales al protoflagelo, lo que produce un flagelo quimiotáctico (6); la fusión de proto-FliN y CheC produce FliM. Cada etapa iría evidentemente seguida de una gradual optimización coevolutiva de las interacciones de los componentes. Así, el origen del flagelo queda reducido a una serie de pasos mutacionales verosímiles.1
Para dar algo
más de
detalle, la siguiente
etapa, si se supone la existencia de un sistema SSTT,
es la
adición de un
filamento. Matzke y muchos otros arguyen que es
fácil elaborar
filamentos
simples de base proteínica —y señalan a
la
polimerización de la hemoglobina en
pacientes de anemia falciforme como resultado de una
sola
mutación puntual
(como cambiar una sola letra en un párrafo y
conseguir una nueva
función). Esta
forma de pensar subestima diversos requisitos muy
específicos
que se precisan
para formar un filamento útil
de
cualquier clase.
Por ejemplo, las partes
de un
filamento
aleatorio, como las que constituyen la hemoglobina
falciforme, tienen
una gran susceptibilidad
de agregarse en amontonamientos o largas hebras
enredadas antes que
sean
transportadas a través de cualquier clase de
poro a la
superficie exterior de
Luego hay una enorme cantidad de otros problemas potenciales para los monómeros filamentosos típicos. ¿Qué de su degradación? ¿Qué del transporte al canal y de la selectividad de la admisión en el canal? ¿Qué del atasco en el interior del canal y del taponamiento del camino? ¿Qué si el filamento acaba formando un núcleo macizo en lugar de un núcleo hueco? ¿Cómo se lograría hacer pasar más piezas filamentosas para añadirlas al extremo distal? ¿Qué sucedería si el extremo no quedase cubierto con una clase diferente de proteína que colocase cada pieza nueva de proteína filamentosa en el lugar apropiado? ¿Cuál es la probabilidad de que cualquier filamento adosado a la maquinaria de exportación vaya a ser «beneficioso» en un medio ambiente determinado —incluso como un «simple» filamento de anclaje?
Ahora bien, no es solo
que las
partes más y más
«especiales» del filamento tienen que
unirse a sí
mismas y de forma correcta,
así como al aparato secretor, sino que tienen
que formar un
filamento cuyo
extremo distal pueda unirse a alguna cosa distinta que
a sí
mismo y a la propia
superficie de la bacteria hospedadora. Por encima de
todo lo
demás, esto parece
que es algo difícil de conseguir.
¿Cuál es la
probabilidad de que un gen capaz
de codificar unas proteínas filamentosas tan
especializadas
llegue a aparecer porque
sí para que sean secretadas de una manera
específica por
un poro de transporte
activo existente?
El
«Simple» Pilus P
A fin de poder siquiera
comenzar a responder a
esta pregunta, consideremos qué se necesita
para elaborar el
más simple
«filamento» bacteriano útil — como
el pilus
P.
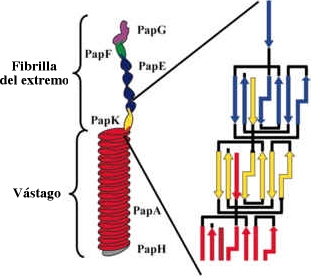
El pilus P funciona como un anclaje de unión entre células bacterianas y otras. Es un delgado filamento hueco ahusado cerca del extremo. Sobre este extremo hay una proteína que se une de forma específica a ciertas clases de moléculas de azúcar en ciertos tipos de células (como células renales). Aunque este pilus es tan simple como se pueda obtener en la vida real e incluso aunque su función parece más bien humilde, está codificado por alrededor de 10 ó 11 genes — tantos como codifican el sistema secretor de tipo III (SSTT) tan evidentemente complejo. La sección proximal más gruesa está formada por componentes proteínicos PapA, y la sección distal más delgada por componentes PapE, y el extremo mismo por PapG (la «adhesina» específica que se une a los azúcares). Hay también una proteína adaptadora, PapF, que une la PapG a la PapE, y otra, PapK, que une la PapE a la PapA.14 Tenemos un total de 5 proteínas diferentes que entran en un orden muy específico. ¿Cómo se consigue este orden?
Este orden se consigue con una interacción bastante complicada de proteínas «chaperonas» o guías. Pero, en primer lugar, la célula tiene que hacer una ruta de exportación multiproteínica llamada ruta sec, que descarga material citoplasmático en el espacio periplasmático. El truco para que las bacterias Gram-negativas «deseen» desarrollar un pilus es hacer penetrar un filamento en la membrana exterior. Esto exige una sofisticada coordinación. En primer lugar, todas las subunidades del pilus se exportan preferentemente, en estado no desplegado, al interior del periplasma a través de la ruta sec donde vuelven a plegarse. Sin embargo, si se dejasen a sí mismas, formarían acumulaciones desorganizadas. De modo que se precisa de una proteína chaperona, la PapD, para impedir este problema de amontonamiento y para ayudar a una conformación apropiada del plegado con el uso de complementación de hebra donante (DSC). Las partes del filamento, por sí mismas, son sumamente inestables y nunca se pliegan de manera apropiada. Y la PapD no tiene ninguna otra función conocida.
A continuación, el
complejo subunidad del pilus-chaperona
interacciona de
manera
específica con un canal proteínico en la
membrana
exterior conocido como PapC.
Este canal es suficientemente grande para que pase a
través suyo
el extremo del
filamento, pero no la parte proximal. La PapD, la
chaperona, entrega la
unidad
del pilus a la PapC, que luego ayuda
en su unión al filamento en crecimiento donde
cada subunidad
contribuye una
hebra para completar de manera perfecta el pliegue de
su vecino, y
así
estabilizarlo.14,15
Así, incluso algo tan relativamente «simple» como la construcción de un pilus parece bastante complicado en comparación con el propuesto paso de la evolución del filamento. Sencillamente, es muy difícil elaborar un filamento «útil» —o así lo parece. Pero digamos que de alguna manera hemos conseguido que evolucione un filamento así. ¿Cómo va luego a evolucionar para llegar a ser un flagelo? Un flagelo necesita secretar proteínas para su construcción. El problema es que no hay ningún pilus P que se sepa que secrete proteínas —quizá debido al pequeño tamaño del canal o a la ausencia de una fuente asociada de energía para bombear las proteínas. En todo caso, todos estos pili son muy diferentes de los flagelos en un aspecto muy importante.
Estos pili se construyen desde el fondo, donde cada nuevo monómero añadido empuja el pilus existente hacia arriba y afuera. Los flagelos, en cambio, son construidos desde el extremo hacia afuera, donde cada nuevo monómero es añadido al extremo de modo que el extremo crece hacia afuera sobre el flagelo existente.
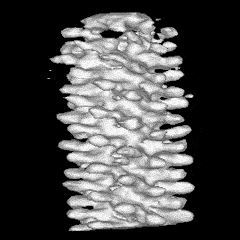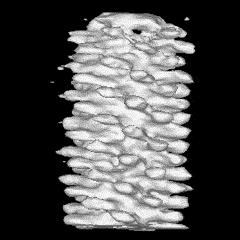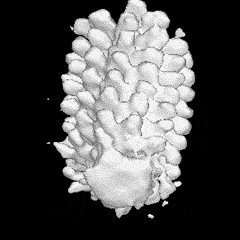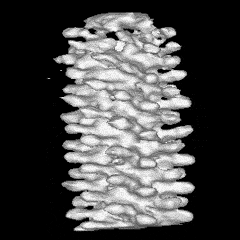
Imagen con
electrón
microscópico del
extremo distal del filamento flagelar
Imagen que aparece en el Proyecto de
Nanomáquinas
Protónicas del
gobierno del Japón: http://www.fbs.osaka-u.ac.jp/labs/namba/npn/movies/FH2gifani.gif
{kind=link}
El desaparecido Robert Macnab,
que fue profesor
de biofísica y bioquímica molecular en
la Universidad de
Yale y que también
estudió los flagelos, observó que el
mecanismo de
construcción flagelar es «un
proceso mucho más sofisticado que lo que
ninguno de nosotros
hubiéramos podido
imaginar».8 Luego
añadió: «Nos
parece que no sería posible que el
sistema funcionase con una complejidad
significativamente menor».9
El
filamento flagelar
Hay un tipo interesante de bacteria sin motilidad, conocida como Shigella, que posee genes flagelares, pero que no produce flagelos. Algunas estirpes de Shigella tienen más genes ausentes que otras, pero en ciertas estirpes el único gen ausente es el gen de FliD. Este gen de FliD codifica la vital proteína de la cubierta del filamento. Sin la proteína de
Detalle
de la cubierta de la flagelina -
Imagen que aparece en el Proyecto de
Nanomáquinas
Protónicas del
gobierno del Japón:
http://www.fbs.osaka-u.ac.jp/labs/namba/npn/movies/CapZoomUp.gif
{kind=link}
La cubierta FliD

Funcionamiento de la
cubierta al ir
alargándose el filamento
Imagen
que
aparece en el
Proyecto de Nanomáquinas Protónicas
del gobierno del
Japón:
http://www.npn.jst.go.jp/movies/CapWorking.gif
La cubierta tiene 5 patas, pero el extremo del
filamento tiene
5,5 subunidades de flagelina en su circunferencia. De
modo que siempre
hay una
pequeña rendija en un punto entre la cubierta y
el filamento. Es
en
este lugar
que se añade la siguiente subunidad en el
filamento en
crecimiento. Al
añadirse
la siguiente subunidad en el espacio abierto, se imprime
un giro a la
cubierta
de modo que se abre un nuevo espacio adyacente al que se
acaba de
llenar. Así,
mientras la cubierta gira y gira, a 10 rotaciones por
segundo, se van
añadiendo
nuevos monómeros de flagelina (FliC) uno por uno,
50 por segundo.8
Lo que es más
interesante en todo esto es que los
extremos de las subunidades de la flagelina se
despliegan mientras se
desplazan
por el tubo del hueco del filamento. Una de las
razones de ello es que
la
flagelina plegada presenta un gran doblez en medio que
la hace
demasiado grande
para viajar a través del tubo. Por sí
mismas, las
subunidades de flagelina no
pueden plegarse apropiadamente. Así,
En resumen, sin esta
cubierta
sumamente
especializada, las unidades de flagelina no pueden
autoestructurarse en
absoluto para constituir un filamento tan ordenado. Y
ni la
proteína de la
cubierta ni los monómeros de la flagelina
tienen ninguna otra
función celular.
Más allá de esto, ¿cómo
sucede que se
coloca la cubierta en la posición
correcta en el extremo del filamento y que no se
envían
más monómeros de la
cubierta por el tubo una vez ha quedado constituido?
Una vez
más, se precisa de
una chaperona específica para la
construcción de la
cubierta y para la
prevención de incorporaciones inoportunas.
Para contrarrestar este
argumento, se dice que
por cuanto las unidades tubulares proteínicas
flagelares FliL y
FliK no
precisan de cubierta alguna para su correcta
construcción, que
la adición de
una cubierta fue una modificación evolutiva
tardía para
mejorar la velocidad y
¿Y qué de
la
hipótesis de que la cubierta se formó
primero, en la que la cubierta hubiera evolucionado
debido a sus
propiedades
adhesivas, y que fue mejorada por la adicional
evolución de las
proteínas de pilus que
extienden la cubierta
hacia
fuera de la célula? De nuevo,
¿cuánto tiempo
sería necesario para la obtención
de un monómero proteínico flagelar lo
suficientemente
específico para
interaccionar con tal cubierta de una forma tan
compleja?
Las explicaciones de
Matzke no
descienden a mayor
detalle que esto. Si estos pasos evolutivos fuesen tan
fáciles
de salvar, sería
fácil ensayarlos en el laboratorio.
Simplemente, se debe
proceder a eliminar el
gen de
La
motorización del flagelo
Bien, digamos que de
alguna manera
alguna
colonia primitiva de bacterias pudo realmente
evolucionar un sistema
proto-SSTT
y un sistema proto-flagelar/filamental donde cada
sistema era funcional
de
forma independiente de alguna manera beneficiosa. En
este punto, Matzke
argumenta que sería algo muy simple unir estos
dos sistemas para
conseguir la
motilidad flagelar.
Al considerar
esta propuesta,
volvamos un poco atrás. Recordemos que el motor
flagelar se
distribuye en dos
unidades básicas: el estator y el rotor. El
estator está
compuesto de unas
subunidades motA y motB (cada una de
ellas formada por
aproximadamente 300 residuos). El rotor está
compuesto de FliM
(~330aa), FliN
(~130aa), y FliG (~330aa). Los tres componentes del
motor están
involucrados en
la construcción del flagelo. El anillo C
formado por estos
componentes actúa
como una especie de taza de medición que
determina el
tamaño del filamento en
forma de gancho. Lo que sucede es que se unen
aproximadamente 120
monómeros de gancho
a la FliM, la FliN y la FliG, (4 lugares de
unión cada uno).
Cuando quedan
llenos todos los lugares de unión, todos los
monómeros
quedan liberados en el
acto y se forma un segmento de «gancho» de
una longitud
específica. Después que
los monómeros del gancho salen del anillo C,
entra otra
proteína y convierte el
anillo C de un secretor de monómeros de gancho
a un secretor de
monómeros de
flagelina. Hay un cambio en la especificidad del
anillo C respecto a
los
monómeros que acepta.
De
modo que la FliG es importante para el montaje
flagelar en cuanto parece que se precisa de los 200
residuos
N-terminales de la
FliG. De hecho, si se distribuyen los 331aa de la FliG
en segmentos de
10aa
cada uno, las mutaciones de deleción de los
segmentos 11, 13,
16, 17, 20, 21, y
27 dan como resultado una falta de formación
adecuada de flagelo
y
evidentemente de la función de motilidad.
Asimismo, las
bacterias con
mutaciones de 1, 3, 12, 14, 15, 22, 23, y 26 de la
FliG son
completamente «no
flageladas».10
Esto significa que la
pretensión de Matzke de que
la FliG, como parte del complejo protosecretor,
«se retiene solo
para
estabilizar/apoyar el complejo secretor coadaptado y
el anillo FliF, y
[es] por
otra parte vestigial» carece totalmente de
sentido.
Naturalmente,
Por lo que se refiere a
la
función de rotación, la
FliM y la FliN son responsables de conmutar el
movimiento en uno y otro
sentido
—no de la creación efectiva de los pares de
fuerza. Sin embargo,
la FliM y la FliN
siguen siendo necesarias para la construcción
del flagelo.
Consideremos ahora brevemente la FliF (complejo de poros de la membrana del anillo MS central con ~550aa). La FliF no tiene homólogos conocidos fuera de los sistemas SSTT (que se consideran evolucionados a partir del sistema flagelar — no al revés). Incluso dada su existencia protoformal, tratar de elaborar una explicación de cómo un flagelo o filamento pudo haberse adherido al mismo de una manera beneficiosa por azar constituye un verdadero reto. La construcción de los filamentos incluso más simples es bastante complicada, como se ha descrito más arriba. Hay diversas proteínas chaperonas involucradas en el acarreo de monómeros específicos a su emplazamiento justo en el momento preciso y en el plegado y en la unión entre sí de forma específica. La construcción de un pilus aparentemente simple es, en realidad, extremadamente compleja. La construcción de un flagelo hueco en su emplazamiento mediante la adición de monómeros en el extremo distal es extraordinariamente complicada.
Dados estos pocos hechos presentados
hasta
ahora, tengo solo
unas pocas preguntas. Matzke sugiere que FliG no tuvo
que
evolucionar con la FliF como parte
del aparato exportador. ¿Cómo se puede
explicar esto si
la FliG es actualmente
necesaria para la construcción del flagelo? Si
la FliG no
evolucionó con la FliF,
¿no sería entonces necesario no solo que
se uniera
enérgicamente a la FliF de
una manera que superase las fuerzas de cizalladura de
la FliG
giratoria, sino
también de una manera que ayudase en la
construcción del
flagelo? De modo que
no solo la FliG tiene que unirse con la FliF, sino que
además ha
de presentar
especificidad en su lugar para un cierto tipo de
monómero
filamentoso. Esto es,
sin la especificidad para la flagelina de la FliG, no
se forma el
flagelo. Cuando
el flagelo comienza a formarse en la vida real, el
anillo MS (FliF) y
el anillo
C (FliG N-terminal + FliN + FliM) se tienen que formar
primero, o no se
formará
el flagelo. Esto parece ineludible.
Quizá se
dirá que
sería más fácil si la FliG
estuviera
ya unida a la FliF —si originalmente la FliG hubiera
evolucionado con
la FliF.
En tal caso ya existiría especificidad para el
monómero
filamentoso en su lugar
y el filamento flagelar ya podría estar en su
lugar,
¿verdad? Pero, en tal
caso, ¿cómo podrían motA/B unirse
a la FliG de una
manera beneficiosa? Hay una
buena cantidad de residuos muy específicos que
tienen que
alinearse justo en el
orden correcto para que la fuerza motriz
protónica de motA/B se
transfiera a
fuerza de par de la FliG — y esto es además de
la simple
unión preferencial de
motA/B con FliG + FliF, ¿verdad?
De modo que, se mire como
se
mire, se precisa de
algo más que una mera unión FliF-FliG.
¿De
qué serviría la especificidad de la FliG
por la flagelina si no estuviera unida primero a la
FliF? ¿Y de
qué serviría la
especificidad de la FliG por la fuerza motriz
protónica de
MotA/B si no
estuviera primero unida a MotA/B? Esta especificidad
involucraría, con una
certidumbre prácticamente total, unas pocas
diferencias de
posición de residuos
adicionales a partir de las «proto-formas»
originales. Y lo
más probable es que
estas diferencias precisas no fuesen secuencialmente
beneficiosas de
una forma
que la selección natural pudiese impulsarlas
adelante.
Más allá de
todo
esto, no va a darse un grado de
unión susceptible de selección entre la
FliG y la FliF
con solo una o dos
posiciones de residuos correctos en su sitio de entre
las 46 posiciones
de
residuos bastante específicos que se emplean
para unir la FliG
con la FliF en
los flagelos modernos. Para vencer los efectos de
sacudida del
movimiento browniano,
el flagelo tiene que girar muy rápidamente
(~100–300 rotaciones
por segundo
durante 3-4 segundos). Esto
significa
que se tienen que vencer un montón de fuerzas
de inercia y de
cizalladura para
mantener la FliG conectada con la FliF.
Tendría
que haber una cantidad significativa de los 46
residuos de
unión, que funcionan
a manera de enganches, todos a la vez, a fin de vencer
estas fuerzas de
cizalladura
en cualquier grado susceptible de selección. De
hecho, los
experimentos de
deleción sugieren que solo el segmento 4
N-terminal de la FliG
puede soportar
algún cambio significativo sin una completa
pérdida de
Sin embargo, resulta que los genes para la FliF y la FliG están situados exactamente contiguos en el genoma. Ciertas mutaciones de deleción entre la FliG y la FliF resultan en una proteína de fusión, una proteína FliG/FliF unida por un enlace covalente que de hecho funciona bastante bien. Está claro que un enlace covalente es mucho más fuerte que un enlace no covalente, de modo que se elimina la necesidad de docenas de enlaces no covalentes. Aunque la proteína de fusión por enlace covalente no funciona tan bien como el sistema silvestre sin enlace covalente, funciona lo suficientemente bien para conseguir realizar el trabajo.
Debido a esta capacidad de enlazar la FliG con la FliF mediante una unión covalente, sin necesidad de conseguir docenas de secuencias correctas, algunos me han dicho que esto hace fácil conseguir que los dos sistemas de nivel inferior beneficiosos de manera independiente (esto es, el motor y el rotor) se unan entre sí para dar origen al sistema de nivel muy superior de motilidad flagelar. Esto, sencillamente, no es cierto, debido a la necesidad multifuncional de la FliG en ambos sistemas a la vez —como se ha descrito más arriba. En pocas palabras, se mire como se mire se necesita más que un simple enlace FliF-FliG. ¿De qué serviría la especificidad de la FliG para la flagelina si no estuviese enlazada primero con la FliF? ¿Y qué de qué serviría la especificidad de la FliG para la fuerza motriz protónica de motA/B si no estuviera primero unida a motA/B? Esta especificidad involucraría bien de seguro bastantes diferencias adicionales de posiciones de los residuos comenzando con las originales «proto-formas». Y, con toda probabilidad, estas diferencias necesarias no serían secuencialmente beneficiosas de una manera en que la selección natural las pudiera impulsar hacia adelante.
Los experimentos
realizados con
mutaciones de
FliF muestran que un «corto segmento de
C-terminal» de 9
residuos aminoácidos
«fundamentales» es imprescindible para la
«construcción del flagelo».
Obsérvese
que este proceso de construcción tiene lugar en
un momento en el
que el motor
está parado y en que no hay rotación de
FliG. Los autores
siguen diciendo que
«la eliminación o sustitución de
hasta 10
aminoácidos inmediatamente encima de
la región fundamental resultaron en un flagelo
paralizado».11 Esto
parece bastante especificado. Los
autores afirmaron que la eliminación o
sustitución de 10
residuos adicionales resultaron en
la
parálisis
del flagelo. Así, parece que la rotación
flagelar
exige algo estructuralmente específico
además de lo que
exija la formación del
flagelo. Tiene que haber
en su sitio alrededor de 19 residuos de
aminoácidos de
Billones
de billones de años
Un
vacío no
beneficioso de solo un par de docenas de residuos
específicos
necesarios en una
posición específica del genoma puede no
parecer mucho a
primera vista, pero
este vacío necesitaría billones de
billones de
años de tiempo medio para que
una población de todas las bacterias en la
tierra (~1030
individuos)
pudiera salvarlo (véase el cálculo en el
apéndice
más adelante). De hecho,
ni uno solo de los pasos
evolutivos propuestos por Matzke u otros se ha
demostrado como factible
en
ningún experimento de laboratorio. Ni tan solo
uno. Sin la
capacidad de poner a
prueba estas historias en el laboratorio, son
simplemente cuestiones no
susceptibles de falsación y por ello, por
definición, no
tienen apoyo en el
método científico. Puede que a muchos
les parezca
extraño siquiera considerar
esto, pero esta clase de pretensiones acerca de la
evolución de
funciones
complejas, del orden de complejidad que encontramos en
el sistema
flagelar, no
son ciencia en absoluto —no son ni siquiera
teorías. Como mucho
son
proposiciones no ensayadas y quizá imposibles
de someter a
ensayo. Dicho
claramente, estos «cuentos» sobre
evolución flagelar
son sencillamente esto —cuentos
de hadas. Y cuando se examinan con cierto detalle, no
parecen
convincentes ni
siquiera sobre el papel.
Sencillamente,
todo esto parece algo más complicado de lo que
Matzke y otros
científicos
evolucionistas parecen querer enseñar.
Consideremos esta
interesantísima conclusión
de
Lynn Margulis, también señalada en una
interesante
reseña
por William Dembski del trabajo de Matzke:
Lo mismo que un
tentempié de bollería
dulce que entretiene el hambre por un momento pero que
nos priva de
alimentos
más nutritivos, el neodarwinismo satisface la
curiosidad
intelectual con
abstracciones vacías de verdaderos detalles —
metabólicos, bioquímicos,
ecológicos o relativos a la historia
natural» (Acquiring
Genomes, p. 103).13
______________________________________________________________________
Referencias
1. Nicholas
Matzke, Evolution in (Brownian) space: a
model for the origin of the bacterial flagellum,
talkreason.org, 2003
(url:
http://www.talkreason.org/articles/flagellum.cfm)
2.
Anand
Sukhan,
Tomoko Kubori, James Wilson, y Jorge E.
Galán. 2001. Genetic Analysis of Assembly of
the Salmonella
enterica Serovar
Typhimurium Type III Secretion-Associated Needle
Complex. J.
Bacteriology
183: 1159-1167.
3.
Macnab,
R.
M., 1999. The bacterial flagellum: reversible
rotary propeller and type III export apparatus. J
Bacteriology. 181 (23), 7149-7153.
4. He,
S. Y., 1998. Type III protein secretion in plant
and animal pathogenic bacteria. Annual Reviews in Phytopathology. 36, 363-392.
5. Kim,
J. F., 2001. Revisiting the chlamydial type III
protein secretion system: clues to the origin of type
III protein
secretion. Trends Genet.
17 (2), 65-69.
6.
Plano,
G. V., Day, J. B. y Ferracci, F., 2001. Type III export: new
uses for an old pathway. Mol
Microbiol. 40
(2), 284-293.
7.
Nguyen,
L.,
Paulsen,
8.
Macnab,
R.
M., Science 290, p. 2087
9.
Macnab
R.
M.,
Bacteria create natural nanomachines, USA
Today, 2005 (ENLACE) .
10.
May
Kihara,
11. Bjorn
Grunenfelder, Stefanie Gehrig, y Urs Jenal, Role
of the Cytoplasmic C Terminus of the FliF Motor
Protein in Flagellar
Assembly
and Rotation, Journal of Bacteriology,
Mar. 2003, p. 1624–1633 Vol. 185, No. 5 (ENLACE).
12. Todas
las animaciones que se
presentan aquí son el asombroso trabajo de
Keiichi Namba et al.
del ERATO
Protonic NanoMachine Project [Proyecto de
Nanomaquinaria
Protónica] (ENLACE).
13. William
Dembski, Biology in the Subjunctive Mood: A
Response to Nicholas Matzke, personal website, 2003. (ENLACE).
14. Yvonne
M.
Lee, Patricia A. DiGiuseppe, Thomas J. Silhavy, y
Scott J. Hultgren, P
Pilus
Assembly Motif Necessary for Activation of the CpxRA
Pathway by PapE in
Escherichia coli, Journal of Bacteriology, July 2004,
p. 4326-4337,
Vol. 186,
No. 13 (ENLACE).
15. Gracias
en especial a Mike Gene por la excelente
información
proporcionada sobre el
tema de la evolución flagelar en su web (ENLACE
DESACTIVADO).
16. Behe,
Michael (1996).
17. Wikipedia,
Irreducible Complexity, último acceso 1/4/2009
(ENLACE).
18. Miller,
Kenneth R. The Flagellum Unspun: The Collapse of
«Irreducible
Complexity» (ENLACE), con
la
respuesta
aquí (ENLACE).
(Último acceso 1/04/09.)
|
Proteína |
Posición |
Ruta
secretora |
Clase
de
operón |
Tamaño |
Estequiometría (aprox.) |
Función |
|
Componentes
integrales de la membrana |
|
|
|
|
||
|
FliF |
Membrana
interna |
sec |
2 |
552 |
26 |
Rotor/Alojamiento |
|
FlhA |
Centro
del
anillo de FliF |
sec? |
2 |
692 |
2? |
Exportación
de
proteína |
|
FlhB |
Centro
del
anillo de FliF |
sec? |
2 |
382 |
2? |
Control
de
longitud del gancho |
|
FliO |
Centro
del
anillo de FliF |
sec? |
2 |
121 |
1? |
Exportación
de
proteína Tipo III |
|
FliP |
Centro
del
anillo de FliF |
sec? |
2 |
245 |
(1?) |
Exportación
de
proteína Tipo III |
|
FliQ |
Centro
del
anillo de FliF |
sec? |
2 |
89 |
(12?) |
Exportación
de
proteína Tipo III |
|
FliR |
Centro
del
anillo de FliF |
sec? |
2 |
261 |
1? |
Exportación
de
proteína Tipo III |
|
|
|
|
|
|
|
|
|
Componentes
asociados a la membrana |
|
|
|
|
|
|
|
FliI |
Lado
citoplasmático
de la membrana |
--- |
2 |
457 |
(6?) |
Exportación
de
proteína Tipo III |
|
FliH |
Lado
citoplasmático
de la membrana |
--- |
2 |
235 |
(2?) |
Exportación
de
proteína Tipo III |
|
FliJ |
Lado
citoplasmático
de la membrana |
--- |
2 |
147 |
(1?) |
Exportación
de
proteína Tipo III |
|
|
|
|
|
|
|
|
|
Complejo
rotor/conmutador |
|
|
|
|
|
|
|
FliM |
Lado
citoplasmático
de la membrana |
automontaje |
|
|
|
Rotor/conmutador |
|
FliN |
Lado
citoplasmático
de la membrana |
automontaje |
|
|
|
Rotor/conmutador |
|
FliG |
Lado
citoplasmático
de la membrana |
automontaje |
|
|
|
Rotor/conmutador |
|
|
|
|
|
|
|
|
|
Anillos |
|
|
|
|
|
|
|
FlgI |
Pared
celular
de peptidoglicano |
sec |
2 |
365 |
26 |
Casquillo-cojinete |
|
FlgH |
Membrana
externa |
sec |
2 |
232 |
26? |
Casquillo |
|
FlgA* |
Espacio
periplasmático |
sec |
2 |
219 |
? |
Montaje
del
anillo P |
|
|
|
|
|
|
|
|
|
Proteínas
axiales |
|
|
|
|
|
|
|
FliE |
Espacio
periplasmático |
Tipo
III |
2 |
104 |
9? |
Vástago
de
transmisión |
|
FlgJ* |
Espacio
periplasmático |
Tipo
III |
2 |
313 |
5? |
Cubierta
del
vástago (?) |
|
FlgB |
Pared
celular
de peptidoglicano (P) |
Tipo
III |
2 |
138 |
6? |
Vástago
de
transmisión |
|
FlgC |
Pared
celular
de peptidoglicano (P) |
Tipo
III |
2 |
134 |
6 |
Vástago
de
transmisión |
|
FlgF |
Pared
celular
de peptidoglicano (P) |
Tipo
III |
2 |
251 |
6 |
Vástago
de
transmisión |
|
FlgG |
Extracelular |
Tipo
III |
2 |
260 |
26 |
Vástago
de
transmisión |
|
FlgE |
Extracelular |
Tipo
III |
2 |
402 |
~130 |
Junta
universal |
|
FlgD* |
Extracelular |
Tipo
III |
2 |
231 |
5? |
Cubierta
del
gancho |
|
FlgK |
Extracelular |
Tipo
III |
3a |
547 |
11 |
Elemento
de
unión |
|
FlgL |
Extracelular |
Tipo
III |
3a |
317 |
11 |
Elemento
de
unión |
|
FliC |
Extracelular |
Tipo
III |
3b |
498 |
~20000 |
Filamento |
|
FliD |
Extracelular |
Tipo
III |
3a |
468 |
5 |
Cubierta
del
filamento |
|
|
|
|
|
|
|
|
|
Proteínas
motoras |
|
|
|
|
|
|
|
MotA |
Membrana
interior/citoplasma |
sec |
3b |
295 |
32? |
Motor/Estator |
|
MotB |
Pared
interior/
peptidoglicano |
sec |
3b |
308 |
16? |
Estator |
|
Proteína |
Posición |
clase
de
operón |
Estequiometría (aprox.) |
Función |
|
Citoplasmática |
|
|
|
|
|
FlhC |
Citoplasma |
1 |
-- |
Regulador
maestro
para operones de clase 2 |
|
FlhD |
Citoplasma |
1 |
-- |
Regulador
maestro
para operones de clase 2 |
|
FlhE |
Citoplasma |
2 |
-- |
? |
|
FliK |
Citoplasma,
se
une con FlhB |
2 |
-- |
Control
de
la longitud del gancho |
|
FliL |
Citoplasma |
2 |
-- |
? |
|
FliA |
Citoplasma |
2 |
-- |
Factor
sigma
para operones de clase 3 |
|
FlgM |
Citoplasma |
3a |
-- |
Factor
anti-sigma |
|
FlgN |
Citoplasma |
3a |
-- |
Chaperona
específica
de FlgK y FlgL |
|
FliS |
Citoplasma |
3a |
-- |
Chaperona
específica
de FliC |
|
FliT |
Citoplasma |
3a |
-- |
Chaperona
específica
de FliD |
|
|
|
|
|
|
|
Quimiotaxia |
|
|
|
|
|
Proteínas
quimiotácticas
aceptoras de metilos
(MCPs) |
(#
copias/célula) |
|
||
|
aer |
Membrana
interna |
-- |
150 |
Receptor
de
oxígeno |
|
tap |
Membrana
interna |
-- |
150 |
Receptor
de
dipéptidos |
|
tar |
Membrana
interna |
-- |
900 |
Receptor
de
aminoácidos |
|
tsr |
Membrana
interna |
-- |
1600 |
Receptor
de
aminoácidos |
|
trg |
Membrana
interna |
-- |
150 |
Receptor
de
proteínas enlazantes con
azúcares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Transducción
de señales |
|
|
|
|
|
CheW |
Lado
interior
de IM (unido a MCP) |
-- |
3000 |
Une
MCP
a CheA |
|
CheA |
Lado
interior
de IM (unido a MCP) |
-- |
3000 |
Histidin-proteín-kinasa
(HPK) |
|
CheY |
Citoplasma |
-- |
3000–17500 |
Regulador
de
respuesta |
|
Regulación
de respuesta |
|
|
|
|
|
CheZ |
Citoplasma |
-- |
1200 |
Regulador
de
respuesta; fostafasa |
|
CheB |
Citoplasma |
-- |
1700 |
Regulador
de
respuesta; metilestearasa |
|
CheR |
Citoplasma |
-- |
850 |
Metiltransferasa |
Apéndice
2
- BILLONES DE
BILLONES DE AÑOS
Cálculo de «billones de
billones de años»
(Enlace)
Tamaño
de población
Tomemos una población
de bacterias del tamaño
de todas las bacterias que existen actualmente en
toda la Tierra
—alrededor de 1e30
bacterias. Digamos que esta población en
estado estacionario
produce una nueva
generación a un ritmo de 20 minutos y que
tiene una tasa de
mutaciones de 1e-8 por
posición de codón —dado un genoma
por bacteria de 10
millones de codones.
¿Cuánto tiempo necesitaría
una población
así para encontrar una nueva función
beneficiosa al nivel de 1.000 residuos
aminoácidos bastante
especificados?
Ante todo,
¿qué son residuos aminoácidos
«bastante especificados»? Se trata de
una medida de la
flexibilidad de la secuencia
que puede ser tolerada por un sistema
funcionalmente benéfico.
Entre otros
ejemplos tenemos enzimas como la lactasa, la
nilonasa o la penicilinasa
u otras
clases de proteínas funcionalmente
benéficas como el
citocromo C (CytoC) que
ayuda a producir energía como parte de la
cadena de transporte
de electrones en
las mitocondrias. También se incluyen
sistemas que exigen
proteínas múltiples
dispuestas de forma específica y que
trabajan conjunta y
simultáneamente —como
los sistemas giratorios de motilidad flagelar.
Cada uno de estos tipos
de
sistemas funcionales de base proteínica
tiene un cierto grado de
flexibilidad
que se puede tolerar sin una pérdida
completa de la
función en cuestión. Sin
embargo, esta flexibilidad tiene un cierto
límite. Algunos
sistemas funcionales
son muy flexibles, mientras que otros tienen
fuertes constricciones. El
CytoC, en
particular, está más limitado de lo
normal, y por ello
tiene un grado bastante
alto de «especificidad» de
secuencia/estructura.
Complejidad de la
secuencia funcional
Algunos autores, como
Durston et. al., designan estos
límites a la
flexibilidad de la secuencia «complejidad
funcional de
secuencia», o «FSC» por
sus siglas en inglés.1
Según Durston et. al., la
medida de la FSC de la molécula
completa es la suma
total de la FSC medida para cada ubicación
en las secuencias
alineadas en
unidades llamadas «correspondencias» [fits].
El valor máximo por situación de
residuo es 4,32
correspondencias/ubicación
(log2 de 20) y este valor puede
materializarse sólo
si puede
tolerarse justo una opción de residuo de
aminoácido (de
entre un total posible
de 20) en el sitio. El valor más bajo
posible es cero, y este
valor se
materializa en una ubicación determinada si
se pueden tolerar
todas las 20
opciones de residuos en dicha ubicación
(esto es, una
flexibilidad total). En
otras palabras, una secuencia al azar
tendría un valor de
correspondencia de
cero. En el estudio de Durston, el promedio de los
valores de
correspondencia [Fit] por
ubicación
(«Densidad de FSC»]
para las proteínas enumeradas es de
alrededor de 2,2 (tomado de
la Tabla 1).1
Por
ejemplo, si encontramos que
la familia de
proteínas ribosómicas S12 tiene un
valor de
correspondencia [Fit] de [359],
podemos usar las
ecuaciones que hemos expuesto hasta ahora para
predecir que hay
alrededor de 1049
diferentes secuencias de los 121 residuos que
podrían entrar en
la familia de
proteínas ribosómicas S12,
resultando en un objetivo de
búsqueda evolutiva de
aproximadamente un 10–106 por ciento
del espacio de
secuencias de los
121 residuos.
En otras palabras,
un valor de
correspondencia [Fit] de
«359» significfa
que sólo 1 de
cada 2359 secuencias tiene la
funcionalidad que se necesita.
Para
conseguir la proporción de secuencias con
este tipo de
función, multiplíquese
esta fracción por el tamaño total de
la secuencia de
espacio de 121aa o 20121.
Tenemos (1/2359) (20121) =
1049. Otra
forma de
decir esto es que el valor promedio por
ubicación para esta
proteína, o la
«densidad de FSC», es de alrededor de
2,966
correspondencias [Fits] por
ubicación de
residuo. Esto
significa que sólo hay disponibles
alrededor de 2,55 opciones de
residuos de
entre 20 por ubicación.
Bajo esta luz,
consideremos el
trabajo de
académicos como Yockey2-5 sobre
estimaciones de
proporciones de
CytoC en el espacio de secuencias. La
estimación de Yockey para
la cantidad de
secuencias con la función CytoC es de
alrededor de 1e65 para un
espacio de
secuencias de 100aa. Esto asciende a alrededor de
la densidad promedio
de FSC
de las secuencias dadas por Durston de alrededor
de 2,2
correspondencias por
ubicación. Para una secuencia de 100aa con
una densidad de FSC
de 2,2 la
fórmula de Durston produciría (1/2220)(20100)
= 1e63; esto
está muy cerca de la predicción de
Yockey. Los grados de
flexibilidad de
secuencia determinados experimentalmente de forma
directa usando
mutaciones en
cassette por investigadores como Sauer, Olsen,
Bowie, Axe y otros
parecen
confirmar estas estimaciones aproximadas generales
para tasas de
sistemas
individuales.6-9
Así,
¿qué
nos dicen estas proporciones acerca del espacio de
secuencia/estructura? ¿Qué
significan? Estas proporciones tan nimias sugieren
poderosamente que
sistemas
bastante especificados como el CytoC y otros
sistemas parecidos son
relativamente raros en el espacio de secuencias.
Sólo para tener
una cierta
idea, todo el Desierto del Sahara contiene
sólo alrededor de 1e30
granos de arena. ¡Y sólo hay
alrededor de
1e80 átomos en todo el
universo! El espacio de secuencias de
proteínas se vuelve
rápidamente mucho más
grande que muchos universos.
¿Y todo esto,
entonces,
qué significa? Bien,
consideremos la probabilidad de que cualquier cosa
dentro del fondo
genético de
1e30 bacterias que estamos considerando (con las
características
señaladas más
arriba) se encuentre dentro de una diferencia de
residuo de un sistema
determinado de base proteínica con un valor
promedio de FSC de
2,2.
Una densidad de FSC
de 2,2
produciría 1e63 secuencias
proteínicas con un tamaño de 100aa
para una
relación de proteínas con la
función dada en cuestión de
alrededor de 1 en 1e67 (o
1e-67 de entre todo el
tamaño del espacio de secuencias de
alrededor de 1e130). Para un
sistema de un
mínimo de 1000aa con la misma densidad de
FSC la cantidad total
de secuencias
con la función dicha sería de
alrededor de 1e638 —un
número muy, pero que muy
grande. Sin embargo, el tamaño total del
espacio de secuencia a
este nivel es
de alrededor de 1e1301 (es decir, 201000).
Esto lleva a una
proporción global de 1 en 1e663.
Así, la
proporción global de 1e-663 al nivel
de 1000aa es un número mucho, pero que
mucho más
pequeño en comparación con el
nivel de FSC de 1e-67 para 100aa —suponiendo el
mismo grado de
especificidad
global. Una diferencia de casi 600 órdenes
de magnitud no es
realmente ni
siquiera susceptible de comparación.
El
vacío de
la Distancia de Hamming
Así,
¿cuál
es el máximo vacío posible
(Distancia
de Hamming) entre cualquier secuencia de partida
en concreto en nuestro
fondo
genético y una de estas 1e638 secuencias
potencialmente
benéficas? Una
secuencia maximalmente distante es aquella que
tiene todas sus
ubicaciones con
un diferente residuo en comparación con la
secuencia de partida
(esto es,
Máxima Distancia de Hamming).
¿Cuántas de estas
secuencias existen en un
espacio de secuencias de 1000aa? Bien, hay 191000
para un
total de
1e1278. Siendo que la cantidad total de secuencias
objetivo es de
1e638, es
posible que todas las secuencias objetivo pudieran
tener una distancia
maximal
de una secuencia de partida.
La
mínima
distancia probable de Hamming
Ahora vamos a la
cuestión verdaderamente
importante: ¿Cuál es la distancia
probable del
vacío desde una secuencia de
partida determinada hasta la secuencia objetivo
más cercana en
el supuesto de
que no se conozcan las ubicaciones reales de las
1e638 secuencias
objetivo? Es
muy improbable que el punto de partida esté
maximalmente
distante de todos los
objetivos. La probabilidad de que una sola
secuencia de 1000aa
esté a una
distancia maximal es de 19/201000 =
~1e-23. Así, la
probabilidad de
que 1e638 de tales secuencias estén a una
distancia maximal es
de 1e-231e638
—un número de extrema improbabilidad. De
modo que la
probabilidad de que una
secuencia de partida esté más cerca
que la máxima
distancia posible para 1000
diferencias para al menos un objetivo en el
espacio de secuencias es
esencialmente segura. Esto parece prometedor para
el mecanismo
evolutivo de
mutaciones al azar y selección natural ...
¿o
quizá no?
Bien, consideremos la probabilidad de que una secuencia de partida tenga 50 diferencias de residuo respecto de una secuencia objetivo determinada con una ubicación desconocida (es decir, que tenga al menos 950 de 1000 ubicaciones iguales que el objetivo). La probabilidad de tener al menos 950 ubicaciones idénticas de residuos se puede calcular usando la fórmula para probabilidades binomiales. La probabilidad de 950 posiciones de residuos coincidentes de entre 1000 (dejando un vacío de diferencias de 50aa) usa la siguiente fórmula para la probabilidad binomial:
|
|
N! k!(N-k)! |
(pk)(1-p)N-k |
Donde:
N =
cantidad de oportunidades
para que se dé el
acontecimiento x;
k =
cantidad de veces que ocurre
el acontecimiento
x o que se estipula que ha de ocurrir;
p =
probabilidad de que el
acontecimiento x
ocurrirá en cualquier ocasión
determinada; y
q =
la probabilidad de que el
acontecimiento x no
ocurrirá en ninguna ocasión
Sustituyendo con
cantidades:
|
P(950 de 1000) =
|
1000! 950!(1000-950)! |
(0,05950)(1–0,05)1000-950 |
=
~1e-1153
Esta es la
probabilidad de
producir exactamente
950 coincidencias a partir de una secuencia de
1000aa.
Añadiendo los
resultados
para coincidencias para
950 hasta 1000 posiciones resulta en una ligera
mejora de la
probabilidad de
que haya al menos una coincidencia dentro de una
diferencia de 50aa
(esto es,
hasta alrededor de 1e-1152).
En otras palabras,
se
precisaría de unas 1e1152 secuencias
de 1000aa para que una de ellas concordase con las
950 posiciones de
residuos
para conseguir un vacío de una amplitud con
una diferencia de
solo 50aa entre
cualquier punto de partida y un objetivo
determinado en el espacio de
secuencia
a estas proporciones —como media. Sin embargo,
tenemos alrededor de
1e638 secuencias
objetivo. ¿Acaso no se dará,
teniendo una cantidad tan
grande de secuencias
objetivo potenciales, que al menos una de ellas
esté dentro de
una distancia de
Hamming de 50? Bien, desde luego que las
probabilidades mejoran, pero
no de
modo significativo. Tomemos la probabilidad
individual de éxito
para encontrar
una sola secuencia objetivo, 1e-1152, y
multipliquemos esta
probabilidad por la
cantidad de secuencias objetivo potenciales,
1e638, y el resultado
sigue siendo
que la probabilidad es de de 1 en 1e514 ciclos de
1e638 ensayos —esto
es, sigue
siendo esencialmente imposible.
Esto es
prácticamente
como encontrar un átomo
marcado situado en una ubicación
desconocida del universo,
compuesto de
alrededor de 1e80 átomos, al menos 6 veces
seguidas ¡al
primer intento cada
vez! Prácticamente todo el mundo
consideraría tal
improbabilidad como una
absoluta «imposibilidad». Esto hace
que una distancia de
vacío tan pequeña como
una diferencia de 50aa en este caso sea
también esencialmente
imposible.
¡Ah, pero
nuestra
población está constituida por 1e30
bacterias con 1e7 codones cada una. Esto es un
total de 1e37 codones
potencialmente diferentes. Esto equivale a 1e34
secuencias con un
tamaño de
1000aa cada una (esto es, 1e34 puntos de partida).
Esto mejora la
probabilidad,
pero no de manera significativa. El cálculo
es 1e514/ 1e34 =
1e480. Es una
mejora en la probabilidad de éxito porque
se necesitarían
menos secuencias, en
promedio, para conseguir una distancia en el
vacío de una
diferencia de 50aa o
menos. Pero esta «mejora»
difícilmente resuelve el
problema. La improbabilidad
sigue ascendiendo esencialmente a una
imposibilidad.
La
cantidad promedio o esperada
de posiciones homólogas de
residuos (esto es,
la
media) es de 50. Esto produce una distancia
esperada promedio de vacío
de Hamming de alrededor de 950
diferencias de residuo entre una secuencia de
partida determinada de
1000aa y
una secuencia objetivo. Dadas estas cifras y las
probabilidades
calculadas, la
sugerencia de que pudiera existir una distancia de
vacío de una
diferencia de
sólo 50aa entre cualquiera de las 1e34
secuencias de partida y
cualquier
secuencia benéfica «objetivo»
potencial parece
extraordinariamente improbable
—hasta el punto de la imposibilidad
práctica.
¿Cuántos
sistemas
funcionales existen?
El
contraargumento,
naturalmente, es que esta
bajísima proporción sería
únicamente
representativa de un solo tipo de función.
En realidad, muchos sistemas potencialmente
benéficos de
diferentes tipos
existirían a los diversos niveles de
espacios de
secuencias/estructuras con
estos requisitos de umbral estructural
mínimo. Así, la
cantidad de secuencias
potencialmente beneficiosas como
combinación colectiva de todos
los tipos
debería aumentarse significativamente a
cada nivel. ¿Por
qué considerar sólo
secuencias del CytoC o de la lactasa?
¡Podría haber
billones de tipos
desconocidos de secuencias potencialmente
beneficiosas en el espacio de
secuencias/estructuras!
Este
es un argumento
interesante. En tanto que es
cierto que tiene que existir una cantidad bastante
grande de tipos
singulares
de sistemas funcionales beneficiosos al nivel de
1000 residuos bastante
especificados, esta cantidad en realidad no afecta
a la
proporción en un grado
significativo por lo que se refiere al problema
que tenemos planteado
—incluso
si se es muy generoso para imaginar la cantidad
global de todos los
tipos de
secuencias potencialmente beneficiosas en el
potencial del espacio de
secuencias a diversos niveles de complejidad
funcional.
En
un artículo publicado
en 2000, Thirumalai y
Klimov hacen los siguientes y pertinentes
comentarios:
Las
estructuras compactas de
mínima energía (MECSs
por sus siglas en inglés), que exhiben
propiedades semejantes a
las de
proteínas, exigen que los estados
fundamentales tengan residuos
H rodeados de
una gran cantidad de residuos hidrofóbicos
tal como se permite
topológicamente.
... Hay implicaciones del espectacular
descubrimiento de que la
cantidad de
MECSs, con características semejantes a las
proteínas, es
muy pequeño, y que no
aumenta significativamente con el tamaño de
la cadena
polipeptídica.
La
cantidad de secuencias
posibles para una
proteína con N aminoácidos es de 20N
que, para
N = 100, es de
aproximadamente 10130. La cantidad de
pliegues en las
proteínas
naturales, que son estructuras compactas de baja
energía libre,
es
evidentemente mucho menor que la cantidad de
posibles secuencias. ...
La
cantidad de estructuras
proteínicas es muy
inferior a la cantidad de secuencias. Al imponer
unos simples rasgos
genéricos
de las proteínas (baja energía y
compactación)
sobre todas las posibles
secuencias observamos que el espacio de
estructuras es escaso en
comparación
con el espacio de secuencias. Incluso si el
espacio de secuencias crece
exponencialmente con N (la cantidad de residuos
aminoácidos [por
20N],
conjeturamos que la cantidad de estructuras
compactas de baja
energía solo
aumenta como lnN [el logaritmo
natural o el poder al que se tendría que
elevar e (2.718 . . . )
para llegar a N]
. . . La cantidad de secuencias para el que
aparece un pliegue
determinado como
estructura nativa se reduce adicionalmente debido
al doble requisito de
estabilidad y de accesibilidad cinética.
... También
sugerimos que el requisito
de funcionalidad puede reducir aún
más la cantidad de
secuencias con
competencia biológica.10
Esta
cifra resultado del
cálculo es respaldada
por otras numerosas estimaciones de la extremada
rareza de secuencias
aminoácidas viables (es decir, estables) en
el espacio de
secuencias
proteínicas. Según la literatura,
parece haber un acuerdo
general de que la
cantidad probable de «pliegues»
proteínicos estables
es inferior a 10.000.
La
cifra estimada de pliegues
varía ampliamente de
700 a ~8000 en base a diferencias en supuestos y
conjuntos de datos
(Orengo et al. 1994; Zhang y
DeLisi 1998;
Govindarajan et al. 1999; Wolf et al.
2000; Coulson y Moult 2002; X.
Liu et al. 2004). El hecho de que
existan
ya 898 pliegues proteínicos solubles,
según la actual
edición de la base de
datos SCOP (v 1.69) (Murzin et al.
1995), indica que la cantidad excederá en
mucho a 1000.14
Nuestros
resultados sugieren que
hay aproximadamente
4.000 posibles pliegues, algunos tan improbables
que sólo
existen aproximadamente
2.000 pliegues entre las proteínas
naturales.20
Los
dominios de los pliegues
tienen generalmente
una longitud de 50–200 residuos y utilizan una
secuencia
específica de cadenas
laterales para codificar estructuras terciarias
enriquecidas en su
estructura secundaria
con núcleos hidrófobos que quedan
protegidos del solvente
por una superficie
predominantemente hidrófila.15
2,0 x
300aa = valor de
correspondencia [fit] 600. 1 en 2600,
o
1e-180
x 20300 = ~1e120 secuencias
estables/viables por pliegue.
Multiplicando 1e120 por 10.000 pliegues = 1e124 de
secuencias
estables/viables
totales en todo el espacio de secuencia de 300aa.
La razón de
viables frente a
no viables es de 1e24 / 20300 =
~1e-267.
¿Pocas estructuras, pero muchas secuencias?
Aunque
muchas secuencias se
pliegan en la
misma o esencialmente la misma estructura 3D como
una cantidad
absoluta, como cantidad
relativa al tamaño del espacio de
secuencias, es una
proporción muy diminuta.
La razón de esto es porque hay «una
disminución
exponencial observada experimentalmente
en la fracción de proteínas
funcionales con cantidades
crecientes de mutaciones
(Bloom et al. 2005)».11
Nuestra
teoría predice
que para grandes cantidades
de sustituciones la probabilidad de que una
proteína retenga su
estructura
disminuirá exponencialmente
con la
cantidad de sustituciones, donde la severidad de
esta
disminución estará
determinada por las propiedades de la estructura.
... Nuestro trabajo
unifica
observaciones relativas al apiñamiento de
proteínas
funcionales en el espacio
de secuencias. ...» [énfasis
añadido].12
Se ha
demostrado mediante
experimentos que las
proteínas pueden ser sumamente tolerantes a
sustituciones
solitarias; por
ejemplo, un 84% de mutantes de un solo residuo de
la lisozima T4 y el
65% de
los mutantes de un solo residuo del represor lac
resultaron funcionales. Para sustituciones
múltiples, la
fracción de proteínas funcionales
disminuye
aproximadamente de forma
exponencial con la cantidad de sustituciones,
aunque la severidad de
esta
disminución varía entre las
proteínas.12
Pero,
a fin de seguir el
argumento, seamos
abrumadoramente generosos y digamos que la
cantidad de sistemas
beneficiosos singularmente diferentes al nivel de
umbral
de 1000aa, desde la perspectiva de una colonia
bacteriana determinada,
no es
justo de 20ln1000 = ~1e9 o mil
millones, o incluso 10 mil
millones,
o un billón, sino de 100 billones (1e14) de
sistemas objetivo
potencialmente
beneficiosos al mismo nivel de complejidad
funcional. Naturalmente,
dado que el
grado de especificidad ya está dado para
este nivel de umbral
(como se ha
indicado antes), cada uno de estos sistemas
queda representado por 1e707 secuencias
aminoácidas
(donde cada sistema de base proteínica
bastante especificado
tiene un tamaño de
1000aa). ¿Qué haría esto
respecto a la cantidad
total de secuencias
potencialmente beneficiosas en el espacio de
secuencias? Bien, lo
multiplicaría
por 1e14 para dar un total de alrededor de 1e721
en lugar de 1e707
secuencias
beneficiosas. Igualmente, aumentaría la
proporción de
beneficioso frente a no
beneficioso 1e14 veces hasta 1e-580 (a partir de
1e-594).
En
relación con el
problema planteado, es poca la
diferencia entre una proporción de 1e-580
respecto de una
proporción de 1e-594.
La probabilidad de tener una sola secuencia de
partida dentro de
diferencias de
50 residuos de cualquier secuencia objetivo
potencialmente viable sigue
siendo
esencialmente cero a alrededor de 1e-431 (esto es,
1e-1152 x 1e721).
Por esta
razón
las islas de secuencias proteínicas
viables/estables están tan aisladas en los
espacios de
secuencias y devienen
exponencialmente más y más aisladas
con los crecientes
requisitos mínimos de
tamaño y/o de especificidad.
El
efecto básico del
plegado de la proteína capturado por este
modelo es que, al irse
plegando la
cadena, es obligada a tener un interior
(núcleo) y un exterior
(superficie)
claramente definidos determinados por la doble
identidad de sus
residuos. La
hidrofobicidad de pequeñas proteínas
de dominio de
plegado simple tiene su pico
hacia alrededor de la mitad, de modo que
aproximadamente una mitad de
los
residuos se fuerzan hacia el núcleo. Una
menor hidrofobicidad
resulta en
secuencias sin plegado, mientras que una mayor
hidrofobicidad lleva a
la
agregación ...
Los
resultados de este
modelo sugieren que el espacio de secuencias de
las proteínas de
dominio de
plegado simple se reparte entre familias de baja
frustración
mutuamente disimilares
que se pliegan en estructuras nativas mutuamente
disimilares. El
principio por
el que surge esta situación es el requisito
de diseño de
frustración minimal,
que permite el plegado eficiente de secuencias en
sus estructuras
funcionales
(nativas) ...
El
espacio de secuencias como poblado por
familias, donde cada una de ellas se pliega en una
burda estructura
granulada
particular y cada una de ella rodeada de una
corteza de secuencias
crecientemente frustradas ... Esto produce una
barrera de
frustración, es
decir, una región de secuencias frustradas
entre cada par de
familias
frustradas minimalmente. Cualquier ruta mutacional
gradual entre una
familia de
secuencia frustrada minimalmente y otra tiene que
visitar luego una
región de
secuencias lentas o carentes de plegado. ... En el
caso de
proteínas reales,
las secuencias en estas regiones de elevada
frustración tienen
mucha menos
propensión a cumplir requisitos
fisiológicos sobre la
capacidad de plegado. ...
Si el requisito es suficiente, la región
entre ambas familias
quedará
completamente excluida, lo que corta el espacio de
secuencias en partes
estables y separadas de pliegue rápido.
Esto proporciona un
mecanismo para la
partición de la información de
secuencia
proteínica en subconjuntos
evolutivamente estables y bioquímicamente
útiles
(plegables).16
Al
nivel de complejidad
bastante especificado de
1000aa, ¿qué significa un
vacío de una diferencia
de residuos de simplemente
500aa — dados los anteriores cálculos que
exponen que con casi
total seguridad
existirá una distancia de Hamming de al
menos 50 entre una isla
determinada de
secuencias viables, por no hablar de beneficiosas,
y la siguiente isla
más
cercana viable potencialmente beneficiosa en el
espacio de secuencias?
¿Qué
hará esta distancia de vacío al
potencial evolutivo?
Pues
bien, consideremos
que incluso para proteínas mucho más
pequeñas con
tamaños de sólo 18aa y vacíos
con distancias de Hamming de menos que media
docena Cui et
al. sugieren que estas distancias
están esencialmente
más allá
de la posibilidad de ser salvadas mediante
mutaciones génicas.
Las
exploraciones
evolutivas por mutaciones génicas pueden
ser asemejadas a la
difusión. Su
alcance es limitado en un paisaje fragmentado de
mortalidad, porque las
secuencias que pertenecen a diferentes sistemas
(Tabla 1) están
fuera de su
alcance.17
Dados
estos
generosísimos supuestos,
¿cuánto
tiempo se necesitará para introducir estos
50 cambios necesarios
de los
caracteres en al menos una secuencia en una
bacteria de nuestra enorme
población de 1e30 bacterias? —una
población tan
grande como la de todas
las bacterias en la Tierra?
Pues
bien, cada una de
nuestras
secuencias de
partida tiene que investigar un espacio de
secuencias de al menos
1e65/1e34 =
~1e31 secuencias antes de alcanzar el
éxito. Con una tasa de
mutaciones de 1e-8
por codón por generación, cada
secuencia de 1.000 codones
mutará una vez cada 1e5
generaciones. Con un tiempo de generación
de 20 minutos, esto
significa una
etapa mutacional cada 2.000.000 minutos; lo que
equivale a ~ 4
años. De modo
que con esta andadura aleatoria/etapa mutacional
cada 4 años, se
necesitarían
1e31 x 4 = 4e31 años para que al menos un
individuo en toda la
población
tuviera éxito —como media (esto es,
billones de billones de
años).
Una gran población en un punto de partida único
Cuando
he presentado
este problema, algunos han sugerido que si toda la
población
comienza a partir
de un solo punto de partida en lugar de estar
esparcida en diferentes
lugares
en el espacio de secuencias, que entonces
mejorarían las
probabilidades de
cruzar una distancia de Hamming de 50. Por
ejemplo, una distancia de
Hamming de
1 alrededor de una isla de 1000aa es igual a 19
veces 1000, o 19.000
secuencias
dentro de una diferencia de un carácter. Si
el tamaño de
población fuese sólo
1000 secuencias idénticas de 1000aa (es
decir, comenzando todos
en la misma
isla), se precisaría de alrededor de 100
generaciones para que
mutase una de
las secuencias en la población. De modo
que, suponiendo que no
hay mutaciones
repetidas, se necesitarían 19.000 x 100 =
1.900.000 generaciones
para explorar
todas las secuencias inmediatamente adyacentes a
la isla del punto de
partida
(alrededor de 72 años). Sin embargo, si la
población de
partida aumentase a 1
millón, alrededor de 10 de estas secuencias
mutarían cada
generación. Así,
suponiendo la inexistencia de mutaciones
repetidas, se
precisaría de 19.000 /
10 = 1.900 generaciones (algo menos que un mes).
Así,
es evidente que el
aumento del tamaño de la
población puede tener un efecto
espectacular acerca de la
velocidad con la que
se puede investigar una distancia de Hamming
determinada. La
cuestión es: ¿que
tamaño de una población de partida
sería necesario
en una isla determinada para
investigar el espacio dentro de una distancia de
Hamming de 50 en un
lapso de
tiempo razonable? ¿En digamos que 4 mil
millones de años?
Veamos.
La distancia de
Hamming de 50 contiene 1e65 secuencias. La
cantidad de 1000aa
secuencias
contenida por todas las bacterias de la tierra es
de solamente 1e34.
Así, si
todas estas secuencias comenzaron en la misma
isla,
¿cuánto se tardaría en
cubrir una distancia de Hamming de 50? En cada
generación se
explorarían 1e29 secuencias.
Así, 1e65/1e29 = 1e36 generaciones ... o
alrededor de 1e31
años (es decir, más
de 10 millones de billones de billones de
años).
Esto
difícilmente
resuelve
el problema ... especialmente si consideramos que
es mucho más
probable que el
vacío de la distancia de Hamming vaya a ser
de más que
150 en lugar de sólo 50
al nivel de 1000 fsaars.
¿Tenemos
más abarrotado el espacio de secuencias?
Pero,
¿qué hay de estos argumentos, a
menudo
fomentados en grupos de discusión como
Talk.Origins, que
sugieren que el
espacio de secuencias está mucho más
abarrotado de
objetivos potenciales que lo
que se menciona aquí?
Las
secuencias
funcionales no son tan infrecuentes ni aisladas.
Hay experimentos que
demuestran que aproximadamente 1 en 1011
de todas las
proteínas de
secuencia aleatoria tienen actividad ATP-ligante
(Keefe y Szostak
2001), y el
trabajo teórico de H. P. Yockey (1992,
326-330) expone que a
esta densidad
todas las secuencias funcionales están
conectadas por cambios
unitarios de
aminoácidos. Véase archivo
de Talk.Origins.
Es
obvio que la
proporción de objetivos frente a no
objetivos puede aumentar,
muy
espectacularmente, en el espacio de secuencias
simplemente rebajando el
tamaño
mínimo y/o el umbral de especificidad bajo
consideración
—rebajando con ello el
«nivel de complejidad funcional».
Algunos tipos de sistemas
tienen una
especificidad de secuencia o requisitos de
tamaños relativamente
bajos. Una
simple función de enlace —como una
secuencia de proteína
enlazando con una
molécula de ATP, sin ningunos otros
requisitos
específicos de unión o
función,
no parece que exija mucha especificidad de
secuencia. Y esto es obvio
por
demostración experimental directa. Sin
embargo, muchos otros
sistemas
funcionales de base proteínica no
están tan poco
especificados. Se precisa de
mucha más secuenciación
específica así como
tamaño para conseguir las funciones
de nivel más elevado. Estas funciones, es
obvio, serán
mucho menos comunes en
el espacio de secuencias y por ello estarán
mucho más
alejadas, en promedio.
Más
específicamente, no
se observa generalmente que las estimaciones
anteriores derivadas
experimentalmente se basan en proteínas con
unos requisitos
mínimos de tamaño
inferiores a 100 residuos aminoácidos
bastante especificados
(fsaars).
Evidentemente, la proporción de objetivos
potenciales por debajo
del umbral
estructural mínimo de 100 fsaars va a ser
exponencialmente
superior a la
proporción por encima de 1.000 fsaars. Esta
es realmente la
clave para
comprender las estimaciones de las proporciones
que tantas veces se
citan como
se menciona más arriba. Estas proporciones
son sólo para
niveles muy bajos de
complejidad funcional que llevan a mucho menos que
1000 fsaars.
¿Son
los
puntos de partida funcionales más
susceptibles de encontrar
objetivos de alto nivel?
Luego
se da el
argumento de que si se comienza con un genoma
funcional intacto, los
puntos de
partida son más susceptibles de combinarse
juntos para producir
al menos
combinaciones proteínicas estables y que
por ello son mucho
más susceptibles de
acertar también en combinaciones
funcionalmente beneficiosas
—especialmente
cuando se consideran mutaciones de
sobrecruzamiento no homólogas
y de
combinación, así como las mutaciones
génicas. Esta
es una sugerencia
interesante, pero consideremos las probabilidades
de este escenario:
Consideremos
un ejemplo simple,
a modo de
ilustración, involucrando un espacio de
secuencias de 20aa.
¿Cuántas secuencias
estables existen de 20aa? (20ln20)(1/244)(2020)
= ~4e16 de entre 1e26 secuencias.
Ahora
bien,
¿cuántas
combinaciones diferentes de secuencias estables
más
pequeñas hay que pudieran
producir una secuencia de 20aa de ninguna clase?
Por ejemplo,
¿cuántas
combinaciones diferentes de secuencias estables de
10aa existen? La
cantidad
total de secuencias estables de 10aa es 20ln10
x 1/222
x
2010 = ~2e9 secuencias diferentes.
De
modo que la cantidad total
de posibles
combinaciones o permutaciones de 2e9, para incluir
inserciones
así como combos
de fin a fin y combos de diferentes longitudes
(como secuencias de 3aa
+ 7aa),
es de 2e92 x 11 x 10 = 4,4e20
secuencias. Añadamos a
esto la
cantidad de mutaciones de sobrecruzamiento no
homólogas de
secuencias estables
de 20aa (1e16 de ellas) y esencialmente se cubre
todo el espacio de
secuencia
de 20aa (~1e26) —que es mucho mayor que la
cantidad de secuencias
estables de
20aa. En pocas palabras, la cantidad de posibles
combinaciones a partir
de
secuencias viables de 20aa o más cortas
exceden inmensamente en
número a la
cantidad total de secuencias estables de 20aa.
Esto significa que la
inmensa
mayoría de las combinaciones posibles,
incluso si se comienza
con secuencias
viables, que acaban produciendo una secuencia de
20aa no
producirá una
secuencia viable de 20aa. Lo mismo es
esencialmente cierto de un
espacio de
secuencias para 1000aa.
Así,
¿hay alguna
ventaja en comenzar con secuencias viables? En realidad, no ...
Consideremos
un trabajo
publicado por Blanco et al.18
Este estudio involucraba dos proteínas, el
dominio SH3 de la
alfa-espectrina (SH3)
y el dominio B1 de la proteína
estreptocócica G (PG), dos
pequeñas proteínas
disimilares. Los investigadores intentaron crear
proteínas
híbridas para
moverse gradualmente a través del espacio
de secuencias desde la
proteína SH3 a
la PG mediante mutaciones de sobrecruzamiento no
homólogas.
Descubrieron que
las secuencias intermedias no se plegaban en
estructuras estables. Lo
que
descubrieron fue que incluso cuando
añadían la mayor
parte de los residuos de
la PG a la secuencia de la SH3, a la vez que
mantenían los
residuos del núcleo
de la SH3, la proteína no se plegaba para
dar una forma estable.
Sólo cuando se
extrajeron los residuos del núcleo de la
SH3 pudo la secuencia
doblarse a la
forma de PG. Además, la estructura de la
SH3 resultó ser
no plegable incluso
cuando se mutaban solo dos residuos no
pertenecientes al núcleo.
Esto implica
que el plegado es una característica
holista, que exige la
cooperación de los
residuos hidrófobos del núcleo y los
residuos
hidrófilos externos al núcleo
para especificar la singular estructura de
«baja
frustración» de una familia
proteínica determinada. Además,
parece que los residuos
de los núcleos de
diferentes familias proteínicas pueden en
realidad
contrarrestarse entre sí,
produciendo una proteína inestable no
plegada cuando se combinan.
El
conjunto de
secuencias analizadas aquí son
híbridos de las secuencias
de SH3 y de PG y
representan un muestreo más o menos
uniforme de identidades de
secuencias entre
100 y ~10% con cada proteína, pero
sólo aquellas
secuencias muy parecidas a las
proteínas de tipo silvestre tienen pliegues
específicos.18
Demostramos
que la
mutación génica por sí misma
es incapaz de hacer
evolucionar sistemas con
pliegues proteínicos sustancialmente
nuevos. Demostramos
además que incluso el
método de barajado del ADN es incapaz de
hacer evolucionar
pliegues proteínicos
sustancialmente nuevos. Nuestras simulaciones
mediante el método
de Monte Carlo
demuestran que el barajado de ADN no
homólogo de estructuras de
baja energía es
una etapa crucial en la búsqueda del
espacio proteínico.19
Mutaciones
génicas en contraste a Mutaciones por
sobrecruzamiento no
homólogo
Como
ilustración de
esto, consideremos un artículo por Cui et
al. (una de cuyas gráficas aparece a
continuación):
El final del artículo de Cui demuestra que el beneficio proporcionado por las recombinaciones no homólogas disminuye con el aumento de la escabrosidad (observada en «α» en la gráfica que aparece arriba) del paisaje adaptativo; y un paisaje muy escabroso proporciona sólo un beneficio marginal en comparación con un paisaje menos escabroso. El concepto de «escabrosidad» es una situación en la que los bordes de las islas están marcadamente definidos en comparación con islas con una pendiente más suave en el espacio de secuencias. Como se ilustra en la figura más abajo, la pendiente de las «islas» beneficiosas de secuencias agrupadas en el espacio de secuencias cae «exponencialmente». Los autores analizan las implicaciones de su Fig. 6 (arriba) con las siguientes palabras:
Así, en tanto que
hay
desde luego una ventaja
estadística en el caso de las mutaciones de
caracteres
múltiples, en la
inclusión de mutaciones de sobrecruzamiento
así como
mutaciones de cortar y
pegar, cuando se trata de encontrar islas beneficiosas
novedosas en el
espacio
de secuencias (por encima de las mutaciones
génicas), esta
ventaja se hace
menos y menos considerable a niveles crecientemente
superiores de
complejidad
funcional. Consideremos que en el trabajo de Cui el
tamaño
máximo de las secuencias
proteínicas que se consideraba era de solamente
18 aa.
Evidentemente, la
densidad de secuencias y/o estructuras beneficiosas
respecto a no
beneficiosas
a este bajísimo nivel va a ser exponencialmente
mayor en
comparación con un
espacio de secuencia de 1000aa cuando se consideren
sistemas que exigen
al
menos un grado de bastante especificidad (como se ha
definido
más arriba).
Además, en tanto que Cui et al.
dicen
que hay una diferencia «significativa»
entre las mutaciones
génicas y los
sobrecruzamientos, parece que los sobrecruzamientos
son sólo
alrededor de dos
veces mejores que las mutaciones génicas,
incluso cuando el
paisaje tiene una
escabrosidad mínima, para poder encontrar
estructuras novedosas
viables.
Y, tal como resulta
ser, el paisaje adaptativo del espacio de secuencias
deviene
linealmente más y
más «escabroso» con cada larguero
que se sube por la
escala de la complejidad
funcional. De modo que el problema de generar sistemas
novedosos y
viables de
base proteínica sigue siendo tan brutal como
siempre —con
independencia de si
se consideran mutaciones génicas o mutaciones
de caracteres
múltiples de la
clase que sea.
Y, tal como resulta
ser, el paisaje adaptativo del espacio de secuencias
deviene
linealmente más y
más «escabroso» con cada larguero
que se sube por la
escala de la complejidad
funcional. De modo que el problema de generar sistemas
novedosos y
viables de
base proteínica sigue siendo tan brutal como
siempre —con
independencia de si
se consideran mutaciones génicas o mutaciones
de caracteres
múltiples de la
clase que sea.
Cuando
el
paisaje es
escabroso, la cantidad de secuencias explorada por las
mutaciones
génicas solas
es comparable con la explorada por las mutaciones
génicas junto
con
sobrecruzamientos [no homólogos]. Esto se debe
a que las
mutaciones génicas son
más eficaces para encontrar un área de
baja mortalidad a
partir de un punto ya
bien poblado en la vecindad, mientras que cuando el
paisaje es abrupto
lo
probable es que mucha de la descendencia de
sobrecruzamientos acaben en
puntos
de mortalidad elevada.17
1. Kirk
K Durston, David KY Chiu,
David L Abel y Jack T Trevors, «Measuring the
functional sequence
complexity of
proteins», Theoretical Biology and
Medical Modelling 2007, 4:47
2. Yockey,
H.P. Information Theory and Molecular
Biology.
3. Yockey, H.P., On
the
information content of cytochrome C, Journal
of
Theoretical Biology, 67 (1977), p. 345-376.
4. Yockey, H. P.
«A Calculation
of the Probability of Spontaneous Biogenesis by
Information
Theory», Journal of Theoretical
Biology (1978)
67, 377-398.
5.
Yockey,
H. P.
«Self
Organization Origin of Life Scenarios and Information
Theory», Journal of Theoretical
Biology (1981)
91, 13-31.
6. Bowie,
J. U., & Sauer, R.
T. (1989) «Identifying Determinants of Folding
and Activity for a
Protein of
Unknown Structure», Proceedings of
the
7.
8. Reidhaar-Olson,
J. F., &
Sauer, R. T. (1990) «Functionally Acceptable
Substitutions in
Two-Helical
Regions of Repressor», Proteins:
Structure, Function, and Genetics 7, 306-316.
9. R.T. Sauer,
James U Bowie,
John F.R. Olson, y Wendall A. Lim, 1989, Proceedings
of
the National Academy of Sciences EE. UU. 86,
2152-2156, y 1990,
16 marzo, Science, 247; y, Olson y
R.T. Sauer, Proteins:
Structure, Function and Genetics,
7:306 - 316, 1990.
10. Thirumalai, D.;
Klimov, D. K., Emergence of stable and fast
folding protein
structures, STOCHASTIC DYNAMICS AND PATTERN FORMATION
IN BIOLOGICAL AND
COMPLEX
SYSTEMS: The APCTP Conference. AIP
Conference
Proceedings, Volumen 501, pp. 95-111 (2000). (ENLACE).
11. Jesse
D. Bloom, Alpan Raval, y
Claus O. Wilke, Thermodynamics of Neutral Protein
Evolution, Genetics. Enero 2007; 175(1):
255–266. (url:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1775007)
12. Jesse
D. Bloom, Jonathan J.
Silberg, Claus O. Wilke, D. Allan Drummond, Christoph
Adami, y Frances
H.
Arnold, Thermodynamic prediction of protein
neutrality, Proc
Natl Acad Sci EE. U.. 18
enero 2005;
102(3): 606–611. (ENLACE)
13. Christina Toft y
Mario A.
Fares, The Evolution of the Flagellar Assembly Pathway
in Endosymbiotic
Bacterial Genomes, Molecular Biology and
Evolution 2008 25(9):2069-2076 (ENLACE).
14. Amit
Oberai, Yungok Ihm,
Sanguk Kim, y James U. Bowie, A limited universe of
membrane protein
families
and folds, Protein Sci. Julio
2006; 15(7): 1723–1734. (ENLACE).
15. J.S.
Richardson, The anatomy
and taxonomy of protein structure. Adv.
Protein Chem. 34 (1981), pp. 167–339. (ENLACE).
16. Erik
David Nelson y Jose
Nelson Onuchic, Proposed mechanism for stability of
proteins to
evolutionary
mutations, Evolution, Vol. 95, pp.
10682–10686, septiembre 1998 (ENLACE).
17. Yan Cui, Wing
Hung Wong, Erich
Bornberg-Bauer, Hue Sun Chan, Recombinatoric
exploration of novel
folded
structures: A heteropolymer-based model of protein
evolutionary
landscapes. PNAS, Vol. 99, Número 2, 809-814, 22 enero
2002. (ENLACE).
18. Francisco J.
Blanco, Isabelle
Angrand y Luis Serrano, Exploring the conformational
properties of the
sequence
space between two proteins with different folds: an
experimental study, Journal of Molecular
Biology, Volumen
285, Número 2, 15 enero 1999, Pp. 741–753 (ENLACE)
19. Bogarad
y Deem, A hierarchical
approach to protein molecular evolution, PNAS Vol. 96,
Número 6,
2591-2595, 16
marzo 1999. (ENLACE).
20. Govindarajan S,
Recabarren R,
Goldstein RA., Estimating the total number of protein
folds, Proteins. 1 junio 1999;
35(4):408-14.
(ENLACE).
Título: La
evolución del
flagelo —y la
ascensión al «Monte Improbable»
Título original: The Evolution of the
Flagellum
—And the
Climbing of "Mt. Improbable"
Autor: Sean D. Pitman,
M.D.
Fuente: http://www.detectingdesign.com/flagellum.html, con permiso. La
versión
castellana
corresponde a la de 2005. Actualización
española de 2009,
correspondiente a la revisión inglesa de enero
de 2009. Para el
original inglés, se puede acudir a la
página de origen de Sean D. Pitman, http://www.detectingdesign.com/flagellum.html
Traducción
del
inglés:
Santiago Escuain
© Copyright 2005, 2009, SEDIN - todos los
derechos reservados.
SEDIN-Servicio
Evangélico
Apartat 2002
08200 SABADELL
(Barcelona) ESPAÑA
Se puede reproducir en todo o en parte para usos no
comerciales, a
condición de que se cite la procedencia
reproduciendo
íntegramente lo anterior y esta nota.
Índice:
Página
principal
Índice
general castellano
Libros recomendados
orígenes
vida
cristiana
bibliografía
general
Coordinadora
Creacionista
Museo
de Máquinas Moleculares
Temas
de actualidad
Documentos
en PDF
(clasificados por temas)
Para descargar la versión en PDF lista para su impresión, haga clic AQUÍ
(Versión actualizada
abril 2009)




||| General English Index ||| Coordinadora Creacionista ||| Museo de Máquinas Moleculares ||| ||| Libros recomendados ||| orígenes ||| vida cristiana ||| bibliografía general ||| ||| Temas de actualidad ||| Documentos en PDF (clasificados por temas) ||| |