|
Servicio
Evangélico de Documentación e
Información
línea sobre línea |
|||||||||| Apartado 2002 - 08200 SABADELL (Barcelona) ESPAÑA | SPAIN ||||||||
¿Es
la resistencia de las bacterias a los antibióticos
|
|
Antibiótico |
Fenotipo que proporciona la resistencia |
|
Actinonina |
Pérdida de actividad enzimática |
|
Ampicilina |
Respuesta SOS que detiene la división celular |
|
Azitromicina |
Pérdida de una proteína reguladora |
|
Cloranfenicol |
Reducción de la formación de una porina o de una proteína reguladora |
|
Ciprofloxacina |
Pérdida de una porina o pérdida de una proteína reguladora |
|
Eritromicina |
Reducción de afinidad a ARNr 23S o pérdida de una proteína reguladora |
|
Fluoroquinolonas |
Pérdida de afinidad a la girasa |
|
Imioenema |
Reducción de la formación de una porina |
|
Kanamicina |
Reducción de la formación de una proteína de transporte |
|
Ácido nalidíxico |
Pérdida o desactivación de una proteína reguladora |
|
Rifampina |
Pérdida de afinidad a la ARN-polimerasa |
|
Estreptomicina |
Afinidad reducida al ARNr 16S o reducción de la actividad de transporte |
|
Tetraciclina |
Formación reducida de una porina o de una proteína reguladora |
|
Zwittermicina A |
Pérdida de fuerza motriz del protón |
La resistencia espontánea a las fluoroquinolonas (como la
ciprofloxacina o la norfloxacina) es también una mutación frecuente en algunas
bacterias. La diana primaria del antibiótico es el enzima ADN-girasa, que está
formado por dos proteínas codificadas por los genes gyrA y gyrB (Hooper y Wolfson, 1993).
El análisis genético ha descubierto que la resistencia a esta clase de
antibióticos puede ser resultado de una mutación puntual en cualquiera de estos
genes (Barnard y Maxwell, 2001; Griggs et al., 1996; Heddle y Maxwell, 2002;
Heisig et al., 1993, Willmott y Maxwell, 1993). Estas mutaciones de las
subunidades de la girasa parecen ser causa de un cambio de conformación
suficiente de la girasa de modo que reduce o pierde su afinidad por las
fluoroquinolonas (Figura 1). Una vez más, a pesar de su naturaleza
«beneficiosa», estas mutaciones no proporcionan un modelo útil que explique el
origen de la afinidad de la girasa por las fluoroquinolonas.
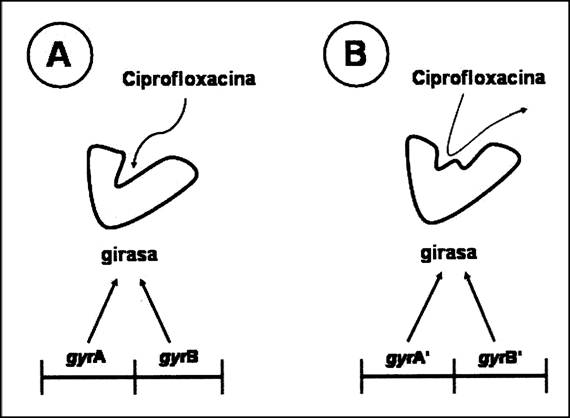
Figura 1. Mecanismo de la resistencia a la ciprofloxacina. (A) La ciprofloxacina interactúa con la girasa, e inhibe su actividad enzimática. (B) Una mutación en cualquiera de ambos genes, gyrA o gyrB, puede cambiar la estructura que conforma la girasa y reducir la afinidad del enzima por la ciprofloxacina. Esto resulta en una incapacidad del antibiótico para inhibir la girasa, y la célula se vuelve resistente al antibiótico.
También la resistencia a la estreptomicina puede proceder de
mutaciones bacterianas espontáneas. En este caso, la estreptomicina bloquea la
síntesis de proteína de la bacteria aparentemente uniéndose con el segmento del
ARNr 16S del ribosoma e interfiriendo con la actividad del ribosoma (Carter et
al., 2000; Leclerc et al., 1991). La resistencia al antibiótico puede surgir por
mutaciones en el gen ARNr 16S gen, que reduce la afinidad de la estreptomicina
para la molécula 16S (Springer et al., 2001). La reducción de unas actividades
de transporte específicas de oligopéptidos lleva también a una resistencia
espontánea frente a diversos antibióticos, incluyendo la estreptomicina
(Kashiwagi et al., 1998). En estos ejemplos, la resistencia surgió como
resultado de la pérdida de un componente o actividad funcionales.
La
pérdida de actividad enzimática puede dar como resultado la resistencia al
metronidazol. El metronidazol intercelular tiene que ser activado mediante
enzimas antes que pueda servir como agente antimicrobiano. Esta activación se
consigue mediante el enzima nitrorreductasa NADPH (Figura 2). Si el metronidazol
no es activado no ejerce un efecto inhibidor sobre la bacteria. Por ello, si no
hay actividad de nitrorreductasa NADPH en la célula, el metronidazol permanece
inactivo. Puede haber pérdida de la actividad de la reductasa por mutaciones
terminadoras o de deleción en rdxA (Debets-Ossenkopp et al.,
1999; Goodwin et al.,
1998; Tankovic et al., 2000). Además, la actividad
de la nitrorreductasa NADPH se puede reducir a causa de una sola mutación de
aminoácido (un solo cambio aminoácido), que reduce su capacidad para activar el
metronidazol (Paul et al., 2001). Todas estas mutaciones resultan en la pérdida
de la actividad enzimática necesaria para que el fármaco sea efectivo en la
célula, y por ello la célula se vuelve resistente al metronidazol. Pero la
pérdida de actividad enzimática no da ningún ejemplo genético de cómo
«evolucionó» originalmente dicho enzima. Por ello, las mutaciones que
proporcionan resistencia frente al metronidazol no pueden presentarse como
verdaderos ejemplos de «evolución en una cápsula Petri».
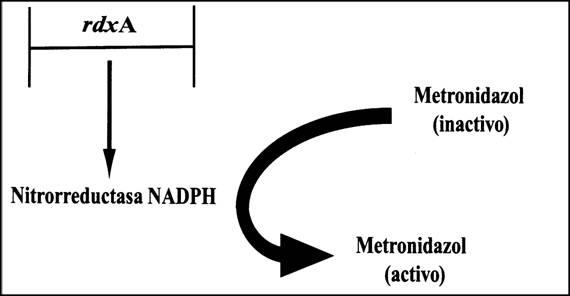
Figura 2. La activación del agente antimicrobiano, el metronidazol. Después de ser transportado al interior de la célula, el metronidazol necesita una modificación estructural para adquirir su forma activa, antimicrobiana. Esta activación se logra por la acción del enzima nitrorreductasa NADPH, que es producto del gen rdxA. Las mutaciones del rdxA pueden impedir la síntesis de una nitrorreductasa NADPH con actividad funcional, lo que impide la activación del metronidazol.
Una diversidad de bacterias, incluyendo la Escherichia
coli, construyen una bomba de eflujo de resistencia múltiple a los
antibióticos (MAR) que proporciona a la bacteria una resistencia a múltiples
tipos de antibióticos, incluyendo la eritromicina, la tetraciclina, la
ampicilina y el ácido nalidíxico. Esta bomba expulsa el antibiótico del
citoplasma de la célula, lo que ayuda a mantener los niveles intracelulares por
debajo de una concentración letal (Grkovic et al., 2002; Okusu et al., 1996)
(Figura 3). La bomba para MAR está compuesta de las proteínas MarA y MarB, la
síntesis de las cuales resulta inhibida por la proteína reguladora, MarR
(Alekshun y Levy, 1999; Poole, 2000) (Figura 3). Las mutaciones que reducen o
eliminan el control de la represión de MarR resultan en una sobreproducción de
la bomba de eflujo MarAB, lo que posibilita a la célula expulsar mayores
concentraciones de antibióticos o de otros agentes bactericidas (Oethinger et
al., 1998; Poole, 2000; Zarantonelli et al., 1999).
La proteína MarA
actúa también como un regulador positivo estimulando una mayor producción de las
proteínas MarA y MarB (Alekshun y Levy, 1999) [Figura 3]. Además, la proteína
MarA inhibe indirectamente la producción de la porina, OmpF, un canal en la
membrana que permite la entrada de algunos antibióticos en la célula (Cohen et
al., 1988). Por ello, la expresión aumentada de MarA aumenta la expulsión de
antibióticos de la célula, y reduce el transporte de algunos antibióticos al
interior de la célula (Figura 3). Las mutaciones de marR que reducen la expresión
o la actividad de la proteína MarR posibilitarán así una expresión excesiva de
la bomba de eflujo MarAB (Linde et al., 2000; Okusu et al., 1996), y
proporcionarán una mayor resistencia de la bacteria a diversos antibióticos
(Eaves et al., 2004; Hans-Jorg et al., 2000; Notka et al., 2002) [Figura 3]. Los
mutantes defectuosos de MarR presentan también una mayor tolerancia bacteriana a
algunos agentes químicos orgánicos, como el ciclohexano (Aono et al., 1998).
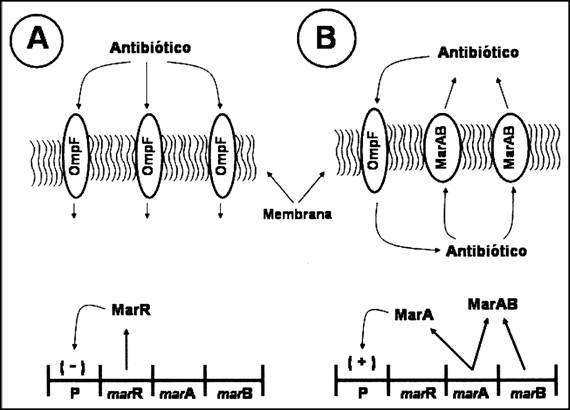
Figura 3. Bomba de eflujo para resistencia a múltiples fármacos. (A) Bacteria sensible a antibióticos. Los antibióticos entran en la célula a través de diversos portales, incluyendo la porina OmpF. La expresión del gen marP produce la proteína reguladora, MarR. Esta proteína se une al promotor (rotulado como P) del operón de resistencia múltiple a los fármacos, inhibiendo la expresión de los genes marA y marB. (B) Bacteria resistente a los antibióticos. Una mutación de marR que que reduce la actividad de MarR hace posible que el promotor funcione constitutivamente. Tanto marA y marB se expresan ahora. Estas dos proteínas forman una bomba de eflujo, que transporta las moléculas de antibiótico fuera del citoplasma de la célula. MarM también se une al promotor (rotulado como P) y aumenta la velocidad de transcripción del operón, lo que aumenta la producción tanto de MarA como de MarB. Además, la producción de MarA reduce de forma indirecta la síntesis de la porina OmpF, con lo que se reduce la cantidad de estas porinas en la membrana, La combinación de un número inferior de porinas para el transporte de un antibiótico al interior de la célula, y el aumento de la cantidad de bombas de eflujo que eliminan el antibiótico de la célula, proporciona a la bacteria una mayor tolerancia a diversos antibióticos.
En otros ejemplos, la resistencia a la eritromicina puede también originarse debido a la pérdida de un segmento de once pares de bases del gen ARNr 23S (Douthwaite et al., 1985), o por una mutación que altera la conformación del ARNr 23S—lo que reduce la afinidad del ribosoma hacia el antibiótico (Gregory y Dahlberg, 1999; Vannuffel et al., 1992). La resistencia al cloranfenicol se obtuvo por deleción de una región de 12 pares de bases en el dominio II del gen de la peptidiltransferasa (Douthwaite, 1992). La resistencia a las cefalosporinas se ha vinculado con una gran alteración de la cinética del transporte en membranas que es semejante a las estirpes deficientes en porinas (Chevalier et al., 1999). La resistencia a la actinonina en el Staphylococcus aureus resulta de mutaciones que eliminan la expresión del gen fmt (Margolis et al., 2000). La resistencia a la zwittermicina A en la E. coli está asociada con la pérdida de fuerza motriz protónica (Stabb y Handelsoman, 1998). En el caso del Streptococcus gordonii, la tolerancia a la penicilina puede involucrar la pérdida del control regulador del operón arc (Caldelari et al., 2000). Y la E. coli puede sobrevivir a la presencia de las β-lactamas, como la ampicilina, deteniendo la división celular, lo que hace la célula menos sensible al efecto letal del antibiótico (Miller et al., 2004).
Estas mutaciones resistentes que se describen aquí llevan a la pérdida de un sistema biológico preexistente, incluyendo la división celular y la fuerza motriz protónica. Aunque la supervivencia antibiótica sea un fenotipo «beneficioso», estas mutaciones no pueden ser un ejemplo genético de cómo se originó cada uno de estos sistemas. Como tales, no proporcionan ningún medio genético para cumplir las predicciones de «descendencia con modificación».
La resistencia a otros antibióticos, como la kanamicina, puede resultar de la pérdida o reducción de síntesis de una proteína transportadora (OppA) [Kashiwagi et al., 1998]. La resistencia a la ciprofloxacina y a la imipenema pueden resultar, al menos en parte, de una disminución en la formación de la porina de la membrana exterior, OmpF (Armand-Lefèvre et al., 2003; Hooper et al., 1987; Yigit et al., 2002). Un aumento en la resistencia al meropenem y a la cefepima va también asociado a la pérdida de OmpF y de otra porina, OmpC (Yigit et al., 2002). Y el Enterobacter aerogenes puede llegar a hacerse resistente a diversos antibióticos cuando una mutación reduce en gran proporción la conductancia de una porina de membrana (Dé et al., 2001).
Cada una de las resistencias que se describen en el párrafo anterior resulta de la reducción o de la pérdida de un sistema de transporte. Sin embargo, los mecanismos genéticos necesarios para la evolución tendrían que dar cuenta del origen de estos diversos sistemas de transporte. Así, estas mutaciones originadoras de la resistencia a los antibióticos no proporcionan los cambios genéticos precisos para la «descendencia común». Al contrario, son genéticamente incongruentes con las necesidades de la evolución, siendo que cada una de ellas involucra la pérdida de una actividad de transporte preexistente.
Como grupo, las mutaciones asociadas con la resistencia a los antibióticos involucran la pérdida o reducción de una función o actividad celular preexistente, esto es, la molécula diana ha perdido una afinidad hacia el antibiótico, el sistema de transporte de antibióticos ha quedado reducido o eliminado, ha habido reducción o eliminación de un sistema regulador o de una actividad enzimática, etc. (Tabla I). Estas no son mutaciones que puedan dar cuenta del origen de dichos sistemas y actividades celulares. Aunque estas mutaciones pueden ciertamente considerarse como «beneficiosas» para la supervivencia de la bacteria cuando está presente un antibiótico en el medio ambiente, este beneficio tiene lugar a expensas de una función previamente existente. Esto es análogo a eliminar una pared interior de una casa para conseguir un comedor más grande. Aunque este comedor mayor pueda ser deseable (esto es, beneficioso), el mecanismo de derribo de esta pared no puede ofrecerse de manera legítima como un ejemplo de cómo se construyó originalmente esta pared interior. Igualmente, el beneficio de la supervivencia de una mutación es solo una parte de los rasgos genéticos necesarios para que las mutaciones puedan dar la «evolución en una cápsula Petri». Estas mutaciones también pueden proporcionar la base genética para una «descendencia común con modificación». Aunque esto contradice de forma directa las pretensiones hechas por muchos proponentes de la evolución, los datos moleculares acerca de la resistencia a los antibióticos son muy claros.
Estas mutaciones tampoco pueden proporcionar un mecanismo que siga «evolucionando» el nivel de especificidad o de actividad de las proteínas que se necesitan para la normal función celular. Aunque estas mutaciones constituyen unos excelentes ejemplos de adaptación bacteriana, son en realidad lo directamente contrario de los cambio por mutación necesarios para la evolución. Sin embargo, estos son precisamente los ejemplos que los evolucionistas presentan como demostraciones verificables del «cambio evolutivo». Cosa irónica, estas mutaciones son en realidad ejemplos verificables de un modelo creacionista—una complejidad inicial que pasa por mutación a un nivel de mayor simplicidad.
La adquisición espontánea de resistencia a los antibióticos es designada con frecuencia como una «ganancia» de resistencia, pero es más apropiado identificarlo como una pérdida de sensibilidad. Así, la resistencia a los antibióticos es resultado de la pérdida de sistemas previamente existentes en la célula bacteriana. Está claro que estos cambios no proporcionan ningún mecanismo genético para el origen de factores celulares como la especificidad enzimática, la actividad de transporte, la actividad reguladora, o la afinidad de las proteínas. Sin embargo, los evolucionistas afirman repetidamente que las mutaciones proporcionan un mecanismo genético para el origen de la actividad biológica y de una «descendencia común con modificación», y presentan repetidamente los tipos de mutación que se acaban de describir como ejemplos de lo mismo.
Costos en la vitalidad debido a la resistencia a los antibióticos
Aunque las mutaciones que proporcionan resistencia a un
antibiótico se pueden considerar «beneficiosos», a menudo comportan un coste
fisiológico (Andersson y Levin, 1999; Maisnier-Patin et al., 2002). De hecho,
Björkman et al. (2000) llegan a la conclusión de que la mayoría de los tipos de
resistencia a los antibióticos impartirán algún coste biológico al organismo.
Por ejemplo, la resistencia a la rifampina del Mycobacterium tuberculosis
(Billington et al., 1999), de la E. coli (Reynolds, 2000), y
del Staphylococcus aureus
(Wichelhaus et al., 2002) se debían a mutaciones de la ARN-polimerasa
que también redujeron la capacidad relativa de la mayoría de las estirpes
mutantes. Aunque el coste biológico comunicado por estos investigadores no era
por lo general muy grave, era mensurable.
Las mutaciones resultantes en
una resistencia a la claritromicina en el Helicobacter pylori reducen la
capacidad relativa del organismo (Björkholm et al., 2001). La resistencia a
elevados niveles de fluoroquinolona por parte de la Salmonella enterica involucra
mutaciones que imparten un elevado costo biológico al organismo (Giraud et al.,
2003). Y las mutaciones de fusA que proporcionan
resistencia al ácido fusídico al Staphylococcus sp. imponen una
significativa pérdida de «capacidad relativa» (Gustafsson et al., 2003; MacVanin
et al., 2000). La
resistencia a la actinonina por parte del S. aureus va también
acompañada de una grave pérdida de «capacidad» que da como resultado una
disminución considerable en el crecimiento (Margolis et al., 2000). La resistencia de la
E. coli a la
estreptomicina puede reducir enormemente la velocidad de biosíntesis de las
proteínas (Zengel et al.,
1977). Y algunas bacterias suspenden la división celular para
minimizar su sensibilidad a la ampicilina (Miller et al., 2004), lo que
evidentemente reduce la capacidad global del organismo.
Este coste de la
«capacidad relativa» parece variar considerablemente, dependiendo tanto del
organismo como del antibiótico. Pero muchos de los mutantes resistentes que han
sido objeto de estudio, incluyendo algunos de los que se han mencionado
anteriormente, pueden posteriormente eliminar algo o mucho del costo sobre la
capacidad biológica mediante retromutaciones o mutaciones supresoras, que
también estabilizan la mutación (Andersson y Levin, 1999; Lenski, 1998; Massey
et al., 2001). El grado en que una retromutación restaura la capacidad biológica
depende probablemente del emplazamiento de la mutación y de si una sola mutación
puede restaurar algo o todo de la «capacidad» del tipo silvestre.
Es
evidente que la capacidad de algunas estirpes mutantes queda reducida de forma
permanente (algunas veces de forma grave), y los evolucionistas, por lo general,
han pasado por alto estos efectos en su precipitación por promover la
resistencia a los antibióticos como «evolución en la cápsula Petri». De hecho, a
menudo someten a ensayo la capacidad relativa de estos mutantes bajo unos
parámetros de cultivo muy rigurosos, que minimizan la pérdida detectable de
capacidad para una mutación determinada. Por otra parte, la pérdida de capacidad
de algunos mutantes es despreciable (especialmente después de retromutaciones).
De modo que el efecto de la resistencia espontánea sobre la capacidad biológica
bacteriana parece variar de mutante en mutante. Sin embargo, las mutaciones
resistentes imponen desde luego un coste biológico por la pérdida de sistemas y
actividades celulares preexistentes. Este coste biológico no queda compensado
por las retromutaciones o por las mutaciones supresoras. Aunque dichas
mutaciones no siempre presenten niveles detectables de reducción de «capacidad»,
se levantan como la antítesis de la «descendencia común con modificación».
Sumario
Con frecuencia se afirma que la resistencia a los antibióticos y a otros microbicidas es una clara demostración de «evolución en una cápsula Petri». Sin embargo, el análisis de los acontecimientos genéticos que causan esta resistencia revela que no son congruentes con los acontecimientos genéticos necesarios para la evolución (definida como «descendencia común con modificación»). En lugar de esto, la resistencia que resulta de la transferencia horizontal de genes proporciona meramente un mecanismo para la transferencia de genes de resistencia previamente existentes. La transferencia horizontal no proporciona un mecanismo para el origen de estos genes. La mutación espontánea sí que ofrece un potencial mecanismo genético para el origen de estos genes, pero este origen nunca se ha podido demostrar. Al contrario, todos los ejemplos conocidos de adquisición de resistencia a los antibióticos debida a mutación son incongruentes con los requisitos genéticos para la evolución. Estas mutaciones dan como resultado la pérdida de sistemas o actividades celulares preexistentes, como porinas y otros sistemas de transporte, sistemas de regulación, actividades enzimáticas y uniones de proteínas. La resistencia a los antibióticos puede también ocasionar alguna disminución de la «capacidad relativa» (grave en algunos casos), aunque en el caso de muchos mutantes esto quede compensado por una reversión. Sin embargo, el verdadero coste biológico es la pérdida de sistemas y actividades preexistentes. Estas pérdidas nunca quedan compensadas, a no ser que se pierda la resistencia, y no se pueden presentar de forma legítima como ejemplos de un cambio evolutivo verdadero.
Referencias
Andersson, D.I., y B.R. Levin. 1999. The biological cost of antibiotic resistance. Current Opinion in Microbiology 2:489–493.
Aono, R., N. Tsukagoshi, y M. Yamamoto. 1998. Involvement of outer membrane protein TolC, a possible member of the mar-sox regulon, in maintenance and improvement of organic solvent tolerance of Escherichia coli K-12. Journal of Bacteriology 180:938–944.
Alekshun, M.N., y S.B. Levy. 1999. The mar regulon: multiple resistance to antibiotics and other toxic chemicals. Trends in Microbiology 7:410–412.
Armand-Lefèvre, L., V. Le.on-Guibout, J. Bredin, F. Barguellil, A. Amor, J.M. Pagès, y M.-H. Nicolas-Chanoine. 2003. Imipenem resistance in Salmonella enterica serovar wien related to porin loss and CMY-4 ß-lactamase production. Antimicrobial Agents and Chemotherapy 47:1165–1168.
Barlow, M., y B.G. Hall. 2002. Phylogentic analysis shows that the OXA â-lactamase genes have been on plasmids for millions of years. Journal of Molecular Evolution 55:314–321.
Barnard, F.M., y A. Maxwell. 2001. Interaction between DNA gyrase and quinolones: effects of alanine mutations at GryA subunit residues Ser83 and Asp87. Antimicrobial Agents and Chemotherapy 45:1994–2000.
Billington, O.J., T.D. McHugh, y S.H. Gillespie. 1999. Physiological cost of rifampin induced in vitro in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 43:1866–1869.
Björkholm, B., I. Nagaev, O.G. Berg, D. Hughes, y D.I. Andersson. 2000. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287:1479–1482.
Björkholm, B., M. Sjölund, P G. Falk, O.G. Berg, L. Engstrand, y D.I. Andersson. 2001. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proceedings of the National Academy of Science 98:14607–14612.
Caldelari, I., B. Loeliger, H. Langen, M .P. Glauser, y P. Moreillon. 2000. Deregulation of the arginine deiminase (arc) operon in penicillin-tolerant mutants of Streptococcus gordonii. Antimicrobial Agents and Chemotherapy 44:2802–2810.
Carter, A.P., W. M. Clemmons, D.E. Brodersen, R.J. Morgan-Warren, B.T. Wimberly, y V. Ramakrishnan. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interaction with antibiotics. Nature 407:340–348.
Chevalier, J., J.-M. Pagès, y M. Malléa. 1999. In vivo modification of porin activity conferring antibiotic resistance to Enterobacter aerogenes. Biochemical and Biophysical Research Communications 266:248–251.
Cohen, S.P., L. M. McMurry, y S.B. Levy. 1988. marA locus causes decreased expression of OmpF porin in multiple-antibiotic-resistant (Mar) mutants of Escherichia coli. Journal of Bacteriology 170:5416–5422.
Darwin, C. 1936. Origin of Species and The Descent of Man (Modern Library Reprint Edition) Random House, New York.
De, E., A. Baslé, M. Jaquinod, N. Saint, M. Malléa, G. Molle, y J.-M Pagès. 2001. A new mechanism of antibiotic resistance in Enterobacteriaceae induced by a structural modification of the major porin. Molecular Microbiology 41:189–198.
Debets-Ossenkopp, Y.J., R..G.J. Pot, D.J. van Westerloo, A. Goddwin, C.M.J.E. Vandenbroucke-Grauls, D.E. Berg, P.S. Hoffman, y J.G. Kusters. 1999. Insertion of mini-IS605 and deletion of adjacent sequences in the nitroreductase (rdxA) gen causes metronidazole resistance in Helicobacter pylori NCTC11637. Antimicrobial Agents and Chemotherapy 43:2657–2662.
Dillon, L.S. 1978. Evolution: concepts and consequences, C.V. Mosby, St. Louis, MO.
Douthwaite, S. 1992. Functional interactions within 23S rRNA involving the peptidyltransferase center. Journal of Bacteriology 174:1333–1338.
Douthwaite, S., J.B. Prince, y H.F. Noller. 1985. Evidence for functional interaction between domains II and V of 23S ribosomal RNA from an erythromycin-resistant mutant. Proceedings of the National Academy of Science 82:8330–8334.
Enright, M., P. Zawadski, P. Pickerill, y C.G. Dowson. 1998. Molecular evolution of rifampicin resistance in Streptococcus pneumoniae. Microbial Drug Resistance 4:65–70.
Eaves, D.J., V. Ricci, y L.J.V. Piddock. 2004. Expression of acrB, acrF, acrD, marA, and soxS in Salmonella enterica serovar typhimurium: role in multiple antibiotic resistance. Antimicrobial Agents and Chemotherapy 48:1145–1150.
Gale, E.F., E. Cundliffe, P.E. Reynolds, M.H. Richmond, y M.J. Waring. 1981. The Molecular Basis of Antibiotic Action. John Wiley & Sons, New York.
Giraud, E., A. Cloeckaert, S. Baucheron, C. Mouline, y E. Chaslus-Dancla. 2003. Fitness cost of fluoroquinolone resistance in Salmonella enterica serovar Typhimurium. Journal of Medical Microbiology 52:697–703.
Gómez, L.R. 1998. Evolution of bacterial resistance to antibiotics during the last three decades. International Microbiology 1:279–284.
Goodwin, A., D. Kersulyte, G. Sisson, S.J.O.V. van Zanten, D.E. Berg, y P.S. Hoffman. 1998. Metronidzole resistance in Helicobacter pylori is due to null mutations in a gen (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Molecular Microbiology 28:383–393.
Gregory, S.T., y A.E. Dahlberg. 1999. Erythromycin resistance mutations in ribosomal proteins L22 and L4 perturb the higher order structure of 23 S ribosomal RNA. Journal of Molecular Biology 289:827–834.
Griggs, D.J., K. Gensberg, y L.J. Piddock. 1996. Mutations in gyrA gen of quinolone-resistant Salmonella serotypes isolated from humans and animals. Antimicrobial Agents and Chemotherapy 40:1009–1013.
Grkovic, S., M.H. Brown, y R.A. Skurray. 2002. Regulation of bacterial drug export systems. Microbiology and Molecular Biology Reviews 66:671–701.
Gustafsson, I., O. Cars, y D.I. Andersson. 2003. Fitness of antibiotic resistant Staphylococcus epidermidis assessed by competition on the skin of human volunteers. Journal of Antimicrobials and Chemotherapy 52;258–263.
Hans-Jörg, L., F. Notka, M. Metze, B. Kochanowski, P. Heisig, y N. Lehn. 2000. Antimicrobial Agents and Chemotherapy 44:1865–1868.
Heddle, J., y A. Maxwell. 2002. Quinolone-binding pocket of DNAgyrase: role of GryB. Antimicrobial Agents and Chemotherapy 46:1805–1815.
Heisig, P., H. Schedletzky, y H. Falkenstein-Paul. 1993. Mutations in the gyrA gen of a highly fluoroquinolone-resistant clinical isolate of Escherichia coli. Antimicrobial Agents and Chemotherapy 37:696–701.
Hooper, D.C., y J.S. Wolfson. 1993. Quinoline Antimicrobial Agents. ASM Press, Washington, DC.
Hooper, D.C., J.S. Wolfson, E. Y. Ng, y M. N. Swartz. 1987. Mechanisms of action and resistance to ciprofloxacin. American Journal of Medicine 82:4A:12–20.
Johnson, G.B. 2000. The Living World. McGraw-Hill, New York.
Kashiwagi, K., M.H. Tsuhako, K. Sakata, T. Saisho, A. Igarashi, S.O.P. daCosta, y K. Igarashi. 1998. Relationship between spontaneous aminoglycoside resistance in Escherichia coli and a decrease in oligopeptide binding protein. Journal of Bacteriology 180:5484–5488.
Leclerc, D., P. Melancon, y L. Brakier-Gingras. 1991. Mutations in the 915 region of Escherichia coli 16S ribosomal RNA reduce the binding of streptomycin to the ribosome. Nucleic Acid Research 19:3973–3977.
Linde, H.-J., F. Notka, M. Metz, B. Kochanowski, P. Heisig, y N. Lehn. 2000. In vivo increased resistance to Ciprofloxacin in Escherichia coli associated with deletion of the C-terminal part of MarR. Antimicrobial Agents and Chemotherapy 44:1865–1868.
Lenski, R.E. 1998. Bacterial evolution and the cost of antibiotic resistance. International Microbiology 1:265–270.
Levin, M.E., y G. F. Hatfull. 1993. Mycobacterium smegmatis RNA polymerase: DNA supercoiling, action of rifampicin and mechanism of rifampicin resistance. Molecular Microbiology 8:277–285.
MacVanin, M., U. Johanson, M. Ehrenberg, y D. Hughes. 2000. Fusidic acid-resistant EF-G perturbs the accumulation of ppGpp. Molecular Microbiology 37:98–107.
Maisnier-Patin, S., O.G. Berg, L. Liljas, y D.I. Andersson. 2002. Compensatory adaptation to the deleterious effects of antibiotic resistance in Salmonella typhimurium. Molecular Microbiology 46:355–366.
Margolis, P.S., C.J. Hackbarth, D.C. Young, W. Wang, D. Chen, Z. Yuan, R. White, y J. Trias. 2000. Peptide deformylase in Staphylococcus aureus: resistance to inhibition is mediated by mutations in the formyltransferase gen. Antimicrobial Agents and Chemotherapy 44:1825–1831.
Massey, R.C., A. Buckling, y S.J. Peacock. 2001. Phenotypic switching of antibiotic resistance circumvents permanent costs in Staphylococcus aureus. Current Biology 11:1810–1814.
Miller, C., L.E. Thomsen, C. Gaggero, R. Mosseri, H. Ingmer, S.N. Cohen. 2004. SOS response induction by ß-lactams and bacterial defense against antibiotic lethality. Science 305:1629–1631.
Miller, K.R. 1999. Finding Darwin’s God. Harper Collins, New York.
Mortlock, R.P. 1984. Microorganisms as Model Systems for Study-ing Evolution. Plenum Press, New York.
Notka, F., H.-J. Linde, A. Dankesreiter, H.-H. Niller, y N. Lehn. 2002. A C-terminal 18 amino acid deletion in MarR in a clinical isolate of Escherichia coli reduces MarR binding properties and increases the MIC of ciprofloxacin. Journal of Antimicrobial Chemotherapy 49:41–47.
Oethinger, M., I. Podglajen, W.V. Kern, y S.T. Levy. 1998. Overexpression of the marA and soxS regulatory gen in clinical topoisomerase mutants of Escherichia coli. Antimicrobial Agents and Chemotherapy 42:2089–2094.
Okusu, H., D. Ma, y H. Nikaido. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-resistance (Mar) mutants. Journal of Bacteriology 178:306–308.
Patterson, C. 1978. Evolution. Cornell University Press, Ithaca, NY.
Paul, R., S. Postius, K. Melchers, y K.P. Schäfer. 2001. Mutations of the Helicobacter pylori genes rdxA and pbp1 cause resistance against metronidazole and amoxicillin. Antimicrobial Agents and Chemotherapy 45:962–965.
Penrose, E. 1998. Bacterial resistance to antibiotics—a case of un-natural selection. Creation Research Society Quarterly 35:76–83.
Poole, K. 2000. Efllux-mediated resistance to fluoroquinolones in gram-negative bacteria. Antimicrobial Agents and Chemo-therapy 44:2233–2241.
Reynolds, M.G. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471–1481.
Springer, B., Y.G. Kidan, T. Prammananan, K. Ellrott, E. C. Böttger, y P. Sander. 2001. Mechanisms of streptomycin resistance: selection of mutations in the 16S rRNA gen conferring resistance. Antimicrobial Agents and Chemotherapy 45:2877–2884.
Stabb, E.V., y J. Handelsman. 1998. Genetic analysis of zwittermicin A resistance in Escherichia coli: effects on membrane potential and RNA polymerase. Molecular Microbiology 27:311–322.
Snyder, L., y W. Champness. 2003. Molecular Genetics of Bacteria. ASM Press, Washington, DC.
Taniguchi, H. H. Aramaki, Y. Nikaido, Y. Mizuguchi, M. Naka-mura, T. Koga, y S. Yoshida. 1996. Rifampicin resistance and mutation of the rpoB gen in Mycobacterium tuberculosis. FEMS Microbiological Letters 144:103–108.
Tankovic, J., D. Lamarque, J.-C. Delchier, C.-J. Soussy, A. Labigne, y P.J. Jenks. 2000. Frequent association between alteration of the rdxA gen and metronidazole resistance in French and North African isolates Helicobacter pylori. Anti-microbial Agents and Chemotherapy 44:608–613.
Top, E.M., Y. Moënne-Loccoz, T. Pembroke, y C.M. Thomas. 2000. Phenotypic traits conferred by plasmids. In Thomas, C.M. (editor), The Horizontal Gen Pool, pp. 249–285. Harwood Academic, Amsterdam, The Netherlands.
Vannuffel, P., M. Di Giambattista, E.A. Morgan, y C. Cocito. 1992. Identification of a single base change in ribosomal RNA leading to erythromycin resistance. Journal of Biological Chemistry 267:8377–8382.
Wang, G., T. J. M. Wilson, Q. Jiang, y D.E. Taylor. 2001. Spontaneous mutations that confer antibiotic resistance in Helicobacter pylori. Antimicrobial Agents and Chemotherapy 45:727–733.
Wichelhaus, T.A., B. Böddinghaus, S. Besier, V. Schäfer, V. Brade, y A. Ludwig. 2002. Biological cost of rifampin from the perspective of Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 46:3381–3385.
Williams, D.L., L. Spring, L. Collins, L.P. Miller, L.B. Heifets, P.R.J. Gangadharam, y T.P. Gillis. 1998. Contribution of rhoB mutations to development of reifamycin cross-resistance in Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 42:1853–1857.
Willmott, C.J.R., y A. Maxwell. 1993. A single point mutation in the DNA gyrase A protein greatly reduces the binding of fluoroquinolones to the gyrase-DNA complex. Antimicrobial Agents and Chemotherapy 37:126–127.
Yigit, H., G. J. Anderson, J. W. Biddle, C. D. Steward, J. K. Rasheed, L.L. Valera, J.E. McGowan, y F.C. Tenover. 2002. Carbapenem resistance in a clinical isolate of Enterobacter aerogenes is associated with decreased expression of OmpF and OmpC porin analogs. Antimicrobial Agents and Chemotherapy 46:3187–3822.
Zarantonelli, L., G. Borthagaray, E.-H. Lee, y W. M. Shafer. 1999. Decreased azithromycin susceptibility of Neisseria gonorrhoeae due to mtrR mutations. Antimicrobial Agents and Chemotherapy 43:2468–2472.
Zengel, J.M., R. Young, P.P. Dennis, y M. Nomura. 1977. Role of ribosomal protein S12 in peptide chain elongation: analysis of pleitropic, streptomycin-resistant mutants of Escherichia coli. Journal of Bacteriology 129:1320–1329.
Fuente: Creation Research Society Quarterly, Vol. 41(4)318-326. marzo de 2005
![]() De vuelta a la página
principal
De vuelta a la página
principal
Nombre original de fichero: 08 Traidores a la Verdad.rtf - preparado el martes, 7 octubre 1997, 10:53
© SEDIN 1997
SEDIN
Servicio Evangélico - Documentación -
Información
Apartado 2002
08200 SABADELL
(Barcelona) ESPAÑA
Índice:
Índice de
boletines
Índice
de línea
sobre línea
Página
principal
Índice
general castellano
Libros recomendados
orígenes
vida
cristiana
bibliografía
general
Coordinadora
Creacionista
Museo de
Máquinas Moleculares
Temas de
actualidad
Documentos en
PDF
(clasificados por temas)




||| General English Index ||| Coordinadora Creacionista ||| Museo de Máquinas Moleculares ||| ||| Libros recomendados ||| orígenes ||| vida cristiana ||| bibliografía general ||| ||| Temas de actualidad ||| Documentos en PDF (clasificados por temas) ||| |